OBJECTIVES
The student understands the primary mechanisms involved in the short-term regulation of arterial pressure:
![]() Identifies the sensory receptors, afferent pathways, central integrating centers, efferent pathways, and effector organs that participate in the arterial baroreceptor reflex.
Identifies the sensory receptors, afferent pathways, central integrating centers, efferent pathways, and effector organs that participate in the arterial baroreceptor reflex.
![]() States the location of the arterial baroreceptors and describes their operation.
States the location of the arterial baroreceptors and describes their operation.
![]() Describes how changes in the afferent input from arterial baroreceptors influence the activity of the sympathetic and parasympathetic preganglionic fibers.
Describes how changes in the afferent input from arterial baroreceptors influence the activity of the sympathetic and parasympathetic preganglionic fibers.
![]() Diagrams the chain of events that are initiated by the arterial baroreceptor reflex to compensate for a change in arterial pressure.
Diagrams the chain of events that are initiated by the arterial baroreceptor reflex to compensate for a change in arterial pressure.
![]() Describes how inputs to the medullary cardiovascular center from cardiopulmonary baroreceptors, arterial and central chemoreceptors, receptors in skeletal muscle, the cerebral cortex, and the hypothalamus influence sympathetic activity, parasympathetic activity, and mean arterial pressure.
Describes how inputs to the medullary cardiovascular center from cardiopulmonary baroreceptors, arterial and central chemoreceptors, receptors in skeletal muscle, the cerebral cortex, and the hypothalamus influence sympathetic activity, parasympathetic activity, and mean arterial pressure.
![]() Describes and indicates the mechanisms involved in the Bezold–Jarisch reflex, the cerebral ischemic response, the Cushing reflex, the alerting reaction, blushing, vasovagal syncope, the dive reflex, and the cardiovascular responses to emotion and pain.
Describes and indicates the mechanisms involved in the Bezold–Jarisch reflex, the cerebral ischemic response, the Cushing reflex, the alerting reaction, blushing, vasovagal syncope, the dive reflex, and the cardiovascular responses to emotion and pain.
The student comprehends the role of the kidney in long-term arterial pressure regulation:
![]() Describes baroreceptor adaptation.
Describes baroreceptor adaptation.
![]() Describes the influence of changes in body fluid volume on arterial pressure and diagrams the steps involved in this process.
Describes the influence of changes in body fluid volume on arterial pressure and diagrams the steps involved in this process.
![]() Indicates the mechanisms whereby altered arterial pressure alters glomerular filtration rate and renal tubular function to influence urinary output.
Indicates the mechanisms whereby altered arterial pressure alters glomerular filtration rate and renal tubular function to influence urinary output.
![]() Describes how mean arterial pressure is adjusted in the long term to that which causes fluid output rate to equal fluid intake rate.
Describes how mean arterial pressure is adjusted in the long term to that which causes fluid output rate to equal fluid intake rate.
![]() Appropriate systemic arterial pressure is the single most important requirement for proper operation of the cardiovascular system. Without sufficient arterial pressure, the brain and the heart do not receive adequate blood flow, no matter what adjustments are made in their vascular resistance by local control mechanisms. In contrast, unnecessary demands are placed on the heart by excessive arterial pressure. Thus, although dramatic changes in peripheral resistance and cardiac function can and do occur normally during the course of our normal daily activities, mean arterial pressure is maintained within a narrow range and is tightly regulated. The elaborate mechanisms that have evolved for regulating this critical cardiovascular variable are discussed in this chapter.
Appropriate systemic arterial pressure is the single most important requirement for proper operation of the cardiovascular system. Without sufficient arterial pressure, the brain and the heart do not receive adequate blood flow, no matter what adjustments are made in their vascular resistance by local control mechanisms. In contrast, unnecessary demands are placed on the heart by excessive arterial pressure. Thus, although dramatic changes in peripheral resistance and cardiac function can and do occur normally during the course of our normal daily activities, mean arterial pressure is maintained within a narrow range and is tightly regulated. The elaborate mechanisms that have evolved for regulating this critical cardiovascular variable are discussed in this chapter.
Arterial pressure is continuously monitored by various sensors located within the body. Whenever arterial pressure varies from normal, multiple reflex responses are initiated, which cause the adjustments in cardiac output, and total peripheral resistance needed to return arterial pressure to its normal value. In the short term (seconds), these adjustments are brought about by changes in the activity of the autonomic nerves leading to the heart and peripheral vessels. In the long term (minutes to days), other mechanisms such as changes in cardiac output brought about by changes in blood volume play an increasingly important role in the control of arterial pressure. The short- and long-term regulations of arterial pressure are discussed in this chapter.
SHORT-TERM REGULATION OF ARTERIAL PRESSURE
Arterial Baroreceptor Reflex
![]()
![]() The arterial baroreceptor reflex is the single most important mechanism providing short-term regulation of arterial pressure. Recall that the usual components of a reflex pathway include sensory receptors, afferent pathways, integrating centers in the central nervous system (CNS), efferent pathways, and effector organs. As shown in Figure 9–1, the efferent pathways of the arterial baroreceptor reflex are the cardiovascular sympathetic and cardiac parasympathetic nerves. The effector organs are the heart and peripheral blood vessels.
The arterial baroreceptor reflex is the single most important mechanism providing short-term regulation of arterial pressure. Recall that the usual components of a reflex pathway include sensory receptors, afferent pathways, integrating centers in the central nervous system (CNS), efferent pathways, and effector organs. As shown in Figure 9–1, the efferent pathways of the arterial baroreceptor reflex are the cardiovascular sympathetic and cardiac parasympathetic nerves. The effector organs are the heart and peripheral blood vessels.
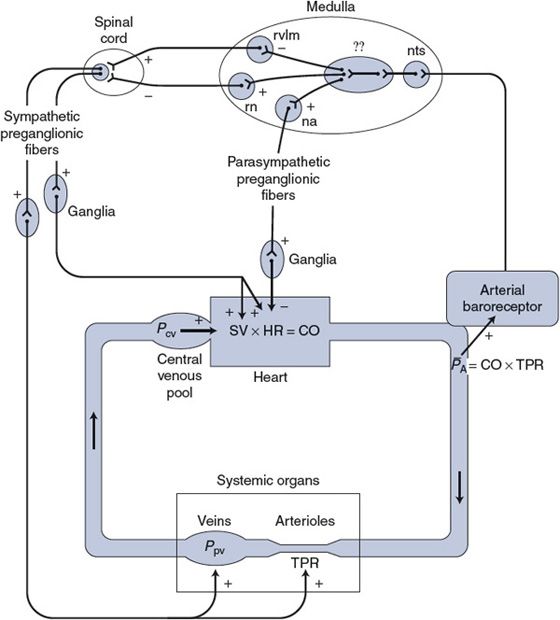
Figure 9–1. Components of the arterial baroreceptor reflex pathway.nts, nucleus tractus solitarius; rvlm, rostral ventrolateral medullary group; rn, raphé nucleus; na, nucleus ambiguus;??, incompletely mapped integration pathways that may also involve structures outside the medulla.
EFFERENT PATHWAYS
![]() Previous chapters have discussed many actions of the sympathetic and parasympathetic nerves leading to the heart and blood vessels. For both systems, postganglionic fibers, whose cell bodies are in ganglia outside the CNS, form the terminal link to the heart and vessels. The influences of these postganglionic fibers on key cardiovascular variables are summarized in Figure 9–1.
Previous chapters have discussed many actions of the sympathetic and parasympathetic nerves leading to the heart and blood vessels. For both systems, postganglionic fibers, whose cell bodies are in ganglia outside the CNS, form the terminal link to the heart and vessels. The influences of these postganglionic fibers on key cardiovascular variables are summarized in Figure 9–1.
The activity of the terminal postganglionic fibers of the autonomic nervous system is determined by the activity of preganglionic fibers whose cell bodies lie within the CNS. In the sympathetic pathways, the cell bodies of the preganglionic fibers are located within the spinal cord. These preganglionic neurons have spontaneous activity that is modulated by excitatory and inhibitory inputs, which arise from centers in the brainstem and descend in distinct excitatory and inhibitory spinal pathways. In the parasympathetic system, the cell bodies of the preganglionic fibers are located within the brainstem. Their spontaneous activity is modulated by inputs from adjacent centers in the brainstem.
AFFERENT PATHWAYS
![]() Sensory receptors, called arterial baroreceptors, are found in abundance in the walls of the aorta and carotid arteries. Major concentrations of these receptors are found near the arch of the aorta (the aortic baroreceptors) and at the bifurcation of the common carotid artery into the internal and external carotid arteries on either side of the neck (the carotid sinus baroreceptors). The receptors themselves are mechanoreceptors that sense arterial pressure indirectly from the degree of stretch of the elastic arterial walls. In general, increased stretch causes an increased action potential generation rate by the arterial baroreceptors. Baroreceptors actually sense not only absolute stretch but also the rate of change of stretch. For this reason, both the mean arterial pressure and the arterial pulse pressure affect baroreceptor firing rate, as indicated in Figure 9–2. The dashed curve in Figure 9–2 shows how baroreceptor firing rate is affected by different levels of a steady arterial pressure. The solid curve in Figure 9–2 indicates how baroreceptor firing rate is affected by the mean value of a pulsatile arterial pressure. Note that in the presence of pulsations (that of course are normal), the baroreceptor firing rate increases at any given level of mean arterial pressure. Note also that changes in mean arterial pressure near the normal value of 100 mm Hg produce the largest changes in baroreceptor discharge rate.
Sensory receptors, called arterial baroreceptors, are found in abundance in the walls of the aorta and carotid arteries. Major concentrations of these receptors are found near the arch of the aorta (the aortic baroreceptors) and at the bifurcation of the common carotid artery into the internal and external carotid arteries on either side of the neck (the carotid sinus baroreceptors). The receptors themselves are mechanoreceptors that sense arterial pressure indirectly from the degree of stretch of the elastic arterial walls. In general, increased stretch causes an increased action potential generation rate by the arterial baroreceptors. Baroreceptors actually sense not only absolute stretch but also the rate of change of stretch. For this reason, both the mean arterial pressure and the arterial pulse pressure affect baroreceptor firing rate, as indicated in Figure 9–2. The dashed curve in Figure 9–2 shows how baroreceptor firing rate is affected by different levels of a steady arterial pressure. The solid curve in Figure 9–2 indicates how baroreceptor firing rate is affected by the mean value of a pulsatile arterial pressure. Note that in the presence of pulsations (that of course are normal), the baroreceptor firing rate increases at any given level of mean arterial pressure. Note also that changes in mean arterial pressure near the normal value of 100 mm Hg produce the largest changes in baroreceptor discharge rate.
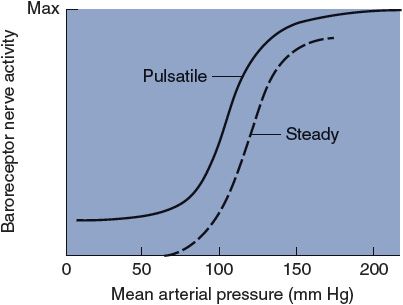
Figure 9–2. The effect of mean arterial pressure on baroreceptor nerve activity.
If arterial pressure remains elevated over a period of several days for some reason, the arterial baroreceptor firing rate will gradually return toward normal. Thus, arterial baroreceptors are said to adapt to long-term changes in arterial pressure. For this reason, the arterial baroreceptor reflex cannot serve as a mechanism for the long-term regulation of arterial pressure.
Action potentials generated by the carotid sinus baroreceptors travel through the carotid sinus nerves (Hering’s nerves), which join with the glossopharyngeal nerves (IX cranial nerves) before entering the CNS. Afferent fibers from the aortic baroreceptors run to the CNS in the vagus nerves (X cranial nerves). (The vagus nerves contain both afferent and efferent fibers, including, for example, the parasympathetic efferent fibers to the heart.)
CENTRAL INTEGRATION
![]() Much of the central integration involved in reflex regulation of the cardiovascular system occurs in the medulla oblongata in what are traditionally referred to as the medullary cardiovascular centers. The neural interconnections between the diffuse structures in this area are complex and not completely mapped. Moreover, these structures appear to serve multiple functions including respiratory control, for example. What is known with a fair degree of certainty is where the cardiovascular afferent and efferent pathways enter and leave the medulla. For example, as indicated in Figure 9–1, the afferent sensory information from the arterial baroreceptors enters the medullary nucleus tractus solitarius, where it is relayed via polysynaptic pathways to other structures in the medulla (and higher brain centers, such as the hypothalamus, as well). The cell bodies of the efferent vagal parasympathetic cardiac nerves are located primarily in the medullary nucleus ambiguus. The sympathetic autonomic efferent information leaves the medulla predominantly from the rostral ventrolateral medulla group of neurons (via an excitatory spinal pathway) or the raphé nucleus (via an inhibitory spinal pathway). The intermediate processes involved in the actual integration of the sensory information into appropriate sympathetic and parasympathetic responses are not well understood at present. Although much of this integration takes place within the medulla, higher centers such as the hypothalamus are probably involved as well. In this context, knowing the details of the integration process is not as important as appreciating the overall effects that changes in arterial baroreceptor activity have on the activities of parasympathetic and sympathetic cardiovascular nerves.
Much of the central integration involved in reflex regulation of the cardiovascular system occurs in the medulla oblongata in what are traditionally referred to as the medullary cardiovascular centers. The neural interconnections between the diffuse structures in this area are complex and not completely mapped. Moreover, these structures appear to serve multiple functions including respiratory control, for example. What is known with a fair degree of certainty is where the cardiovascular afferent and efferent pathways enter and leave the medulla. For example, as indicated in Figure 9–1, the afferent sensory information from the arterial baroreceptors enters the medullary nucleus tractus solitarius, where it is relayed via polysynaptic pathways to other structures in the medulla (and higher brain centers, such as the hypothalamus, as well). The cell bodies of the efferent vagal parasympathetic cardiac nerves are located primarily in the medullary nucleus ambiguus. The sympathetic autonomic efferent information leaves the medulla predominantly from the rostral ventrolateral medulla group of neurons (via an excitatory spinal pathway) or the raphé nucleus (via an inhibitory spinal pathway). The intermediate processes involved in the actual integration of the sensory information into appropriate sympathetic and parasympathetic responses are not well understood at present. Although much of this integration takes place within the medulla, higher centers such as the hypothalamus are probably involved as well. In this context, knowing the details of the integration process is not as important as appreciating the overall effects that changes in arterial baroreceptor activity have on the activities of parasympathetic and sympathetic cardiovascular nerves.
Several functionally important points about the central control of the autonomic cardiovascular nerves are illustrated in Figure 9–1. The major external influence on the cardiovascular centers comes from the arterial baroreceptors. Because the arterial baroreceptors are active at normal arterial pressures, they supply a tonic input to the central integration centers.
![]() As indicated in Figure 9–1, the integration process is such that increased input from the arterial baroreceptors tends to simultaneously (1) inhibit the activity of the spinal sympathetic excitatory tract, (2) stimulate the activity of the spinal sympathetic inhibitory tract, and (3) stimulate the activity of parasympathetic preganglionic nerves. Thus, an increase in the arterial baroreceptor discharge rate (caused by increased arterial pressure) causes a decrease in the tonic activity of cardiovascular sympathetic nerves and a simultaneous increase in the tonic activity of cardiac parasympathetic nerves. Conversely, decreased arterial pressure causes increased sympathetic and decreased parasympathetic activity.
As indicated in Figure 9–1, the integration process is such that increased input from the arterial baroreceptors tends to simultaneously (1) inhibit the activity of the spinal sympathetic excitatory tract, (2) stimulate the activity of the spinal sympathetic inhibitory tract, and (3) stimulate the activity of parasympathetic preganglionic nerves. Thus, an increase in the arterial baroreceptor discharge rate (caused by increased arterial pressure) causes a decrease in the tonic activity of cardiovascular sympathetic nerves and a simultaneous increase in the tonic activity of cardiac parasympathetic nerves. Conversely, decreased arterial pressure causes increased sympathetic and decreased parasympathetic activity.
OPERATION OF THE ARTERIAL BARORECEPTOR REFLEX
The arterial baroreceptor reflex is a continuously operating control system that automatically makes adjustments to prevent primary disturbances on the heart and/or vessels from causing large changes in mean arterial pressure. The arterial baroreceptor reflex mechanism acts to regulate arterial pressure in a negative feedback manner that is analogous in many ways to the manner in which a thermostatically controlled home heating system operates to regulate inside temperature despite disturbances such as changes in the weather or open windows.1
![]() Figure 9–3 shows many events in the arterial baroreceptor reflex pathway that occur in response to a disturbance of decreased mean arterial pressure. All the events shown in Figure 9–3 have already been discussed, and each should be carefully examined (and reviewed if necessary) at this point because a great many of the interactions that are essential to understanding cardiovascular physiology are summarized in this figure.
Figure 9–3 shows many events in the arterial baroreceptor reflex pathway that occur in response to a disturbance of decreased mean arterial pressure. All the events shown in Figure 9–3 have already been discussed, and each should be carefully examined (and reviewed if necessary) at this point because a great many of the interactions that are essential to understanding cardiovascular physiology are summarized in this figure.
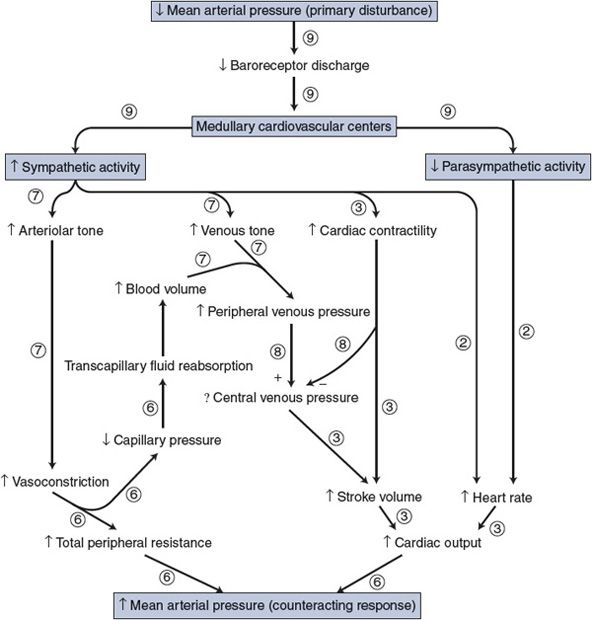
Figure 9–3. Immediate cardiovascular adjustments caused by a decrease in arterial blood pressure. Circled numbers indicate the chapter in which each interaction was previously discussed.
Note that in Figure 9–3 the overall response of the arterial baroreceptor reflex to the disturbance of decreased mean arterial pressure is increased mean arterial pressure (ie, the response tends to counteract the disturbance). A disturbance of increased mean arterial pressure would elicit events exactly opposite to those shown in Figure 9–3 and produce the response of decreased mean arterial pressure; again, the response tends to counteract the disturbance. The homeostatic benefits of the reflex action should be apparent.
One should recall that nervous control of vessels is more important in some areas such as the kidney, the skin, and the splanchnic organs than in the brain and the heart muscle. Thus, the reflex response to a fall in arterial pressure may, for example, include a significant increase in renal vascular resistance and a decrease in renal blood flow without changing the cerebral vascular resistance or blood flow. The peripheral vascular adjustments associated with the arterial baroreceptor reflex take place primarily in organs with strong sympathetic vascular control.
Other Cardiovascular Reflexes and Responses
![]() Seemingly in spite of the arterial baroreceptor reflex mechanism, large and rapid changes in mean arterial pressure do occur in certain physiological and pathological situations. These reactions are caused by influences on the medullary cardiovascular centers other than those from the arterial baroreceptors. As outlined in the following sections, these inputs on the medullary cardiovascular centers arise from many types of peripheral and central receptors as well as from “higher centers” in the CNS such as the hypothalamus and the cortex.
Seemingly in spite of the arterial baroreceptor reflex mechanism, large and rapid changes in mean arterial pressure do occur in certain physiological and pathological situations. These reactions are caused by influences on the medullary cardiovascular centers other than those from the arterial baroreceptors. As outlined in the following sections, these inputs on the medullary cardiovascular centers arise from many types of peripheral and central receptors as well as from “higher centers” in the CNS such as the hypothalamus and the cortex.
The analogy was made earlier that the arterial baroreceptor reflex operates to control arterial pressure somewhat as a home heating system acts to control inside temperature. Such a system automatically acts to counteract changes in temperature caused by such things as an open window or a dirty furnace. It does not, however, resist changes in indoor temperature caused by someone’s resetting of the thermostat dial—in fact, the basic temperature regulating mechanisms cooperate wholeheartedly in adjusting the temperature to the new desired value. The temperature setting on a home thermostat’s dial has a useful conceptual analogy in cardiovascular physiology often referred to as the “set point” for arterial pressure. Most (but not all) of the influences that are about to be discussed influence arterial pressure as if they changed the arterial baroreceptor reflex’s set point for pressure regulation. Consequently, the arterial baroreceptor reflex does not resist most of these pressure disturbances but actually assists in producing them.
REFLEXES FROM RECEPTORS IN THE HEART AND LUNGS
A host of mechanoreceptors and chemoreceptors that can elicit reflex cardiovascular responses have been identified in atria, ventricles, coronary vessels, and lungs. The role of these cardiopulmonary receptors in neurohumoral control of the cardiovascular system is, in most cases, incompletely understood, but they are likely to be importantly involved in regulating blood volume and body fluid balance.
One general function that the cardiopulmonary receptors perform is sensing the pressure (or volume) in the atria and the central venous pool. Increased central venous pressure and volume cause receptor activation by stretch, which elicits a reflex decrease in sympathetic activity. Decreased central venous pressure produces the opposite response. Whatever the details, it is clear that cardiopulmonary baroreflexes normally exert a tonic inhibitory influence on sympathetic activity and play an arguably important, but not yet completely defined, role in normal cardiovascular regulation.2
CHEMORECEPTOR REFLEXES
Low PO2 and/or high PCO2 levels in the arterial blood cause reflex increases in respiratory rate and mean arterial pressure. These responses appear to be a result of increased activity of arterial chemoreceptors, located in the carotid arteries and the arch of the aorta, and central chemoreceptors, located somewhere within the CNS. Chemoreceptors probably play little role in the normal regulation of arterial pressure because arterial blood PO2 and PCO2 are normally held very nearly constant by respiratory control mechanisms.
An extremely strong reaction called the cerebral ischemic response is triggered by inadequate brain blood flow (ischemia) and can produce a more intense sympathetic vasoconstriction and cardiac stimulation than is elicited by any other influence on the cardiovascular control centers. Presumably the cerebral ischemic response is initiated by chemoreceptors located within the CNS. However, if cerebral blood flow is severely inadequate for several minutes, the cerebral ischemic response wanes and is replaced by marked loss of sympathetic activity. Presumably this situation results when function of the nerve cells in the cardiovascular centers becomes directly depressed by the unfavorable chemical conditions in the cerebrospinal fluid.
Whenever intracranial pressure is increased—for example, by tumor growth or trauma-induced bleeding within the rigid cranium—there is a parallel rise in arterial pressure. This is called the Cushing reflex and is a variant of the cerebral ischemic response. It can cause mean arterial pressures of more than 200 mm Hg in severe cases of intracranial pressure elevation. The obvious benefit of the Cushing reflex is that it prevents collapse of cranial vessels and thus preserves adequate brain blood flow in the face of large increases in intracranial pressure. The mechanisms responsible for the Cushing reflex are not fully understood but seem to involve central (CNS) chemoreceptors reacting to consequences of reduced cerebral circulation producing very strong sympathetic activation. A major increase in total peripheral vascular resistance occurs. The early phase of the Cushing reflex often includes tachycardia, whereas the late (and more dangerous) phase of this reflex is accompanied by bradycardia (presumably resulting from elevated reflex vagal activity from the arterial baroreceptor input).
REFLEXES FROM RECEPTORS IN EXERCISING SKELETAL MUSCLE
Reflex tachycardia and increased arterial pressure can be elicited by stimulation of certain afferent fibers from the skeletal muscle. These pathways may be activated by chemoreceptors responding to muscle ischemia, which occurs with strong, sustained static (isometric) exercise. This input may contribute to the marked increase in blood pressure that accompanies such isometric efforts. It is uncertain as to what extent this reflex contributes to the cardiovascular responses to dynamic (rhythmic) muscle exercise.
THE DIVE REFLEX
Aquatic animals respond to diving with a remarkable bradycardia and intense vasoconstriction in all systemic organs except the brain and the heart. The response serves to allow prolonged submersion by limiting the rate of oxygen use and by directing blood flow to essential organs. A similar but less dramatic dive reflex can be elicited in humans by simply immersing the face in water. (Cold water enhances the response.) The response involves the unusual combination of bradycardia produced by enhanced cardiac parasympathetic activity and peripheral vasoconstriction caused by enhanced sympathetic activity. This is a rare exception to the general rule that sympathetic and parasympathetic nerves are activated in reciprocal fashion. The dive reflex is sometimes used clinically to reflexly activate cardiac parasympathetic nerves for the purpose of interrupting atrial tachyarrhythmias.
Another, but unrelated, clinical technique for activating parasympathetic nerves in an attempt to interrupt atrial tachyarrhythmias is called carotid massage. In essence, massage of the neck is done to cause physical deformation of the carotid sinuses and “trick” them into sending a “high-pressure” alarm to the medullary control centers.
CARDIOVASCULAR RESPONSES ASSOCIATED WITH EMOTION
Cardiovascular responses are frequently associated with certain states of emotion. These responses originate in the cerebral cortex and reach the medullary cardiovascular centers through corticohypothalamic pathways. The least complicated of these responses is the blushing that is often detectable in individuals with lightly pigmented skin during states of embarrassment. The blushing response involves a loss of sympathetic vasoconstrictor activity only to particular cutaneous vessels, and this produces the blushing by allowing engorgement of the cutaneous venous sinuses.
Excitement or a sense of danger often elicits a complex behavioral pattern called the alerting reaction (also called the “defense” or “fight or flight” response). The alerting reaction involves a host of responses such as pupillary dilation and increased skeletal muscle tenseness that are generally appropriate preparations for some form of intense physical activity. The cardiovascular component of the alerting reaction is an increase in blood pressure caused by a general increase in cardiovascular sympathetic nervous activity and a decrease in cardiac parasympathetic activity. Centers in the posterior hypothalamus are presumed to be involved in the alerting reaction because many of the components of this multifaceted response can be experimentally reproduced by electrical stimulation of this area. The general cardiovascular effects are mediated via hypothalamic communications with the medullary cardiovascular centers.
Some individuals respond to situations of extreme stress by fainting, a situation referred to clinically as vasovagal syncope. The loss of consciousness is due to decreased cerebral blood flow that is itself produced by a sudden dramatic loss of arterial blood pressure that, in turn, occurs as a result of a sudden loss of sympathetic tone and a simultaneous large increase in parasympathetic tone and decrease in the heart rate. The influences on the medullary cardiovascular centers that produce vasovagal syncope appear to come from the cortex via depressor centers in the anterior hypothalamus. It has been suggested that vasovagal syncope is analogous to the “playing dead” response to peril used by some animals. Fortunately, unconsciousness (combined with becoming horizontal) seems to quickly remove this serious disturbance to the normal mechanisms of arterial pressure control in humans.
The extent to which cardiovascular variables, in particular blood pressure, are normally affected by an emotional state is currently a topic of extreme interest and considerable research. As yet the answer is unclear. However, the therapeutic value of being able, for example, to learn to consciously reduce one’s blood pressure would be incalculable.
CENTRAL COMMAND
The term central command is used to imply an input from the cerebral cortex to lower brain centers during voluntary muscle exercise. The concept is that the same cortical drives that initiate somatomotor (skeletal muscle) activity also simultaneously initiate cardiovascular (and respiratory) adjustments appropriate to support that activity. In the absence of any other obvious causes, central command is at present the best explanation as to why both mean arterial pressure and respiration increase during voluntary exercise.
REFLEX RESPONSES TO PAIN
Pain can have either a positive or a negative influence on arterial pressure. Generally, superficial or cutaneous pain causes a rise in blood pressure in a manner similar to that associated with the alerting response and perhaps over many of the same pathways. Deep pain from receptors in the viscera or joints, however, often causes a cardiovascular response similar to that which accompanies vasovagal syncope, that is, decreased sympathetic tone, increased parasympathetic tone, and a serious decrease in blood pressure. This response may contribute to the state of shock that often accompanies crushing injuries and/or joint displacement.
TEMPERATURE REGULATION REFLEXES
Certain special cardiovascular reflexes that involve the control of skin blood flow have evolved as part of the body temperature regulation mechanisms. Dilation of cutaneous vessels promotes heat loss (as long as the environmental temperature is below the body temperature). Temperature regulation responses are controlled primarily by the hypothalamus, which can operate through the cardiovascular centers to discretely control the sympathetic activity to regulate vasoconstriction of cutaneous vessels and thus skin blood flow. The sympathetic activity to cutaneous vessels is extremely responsive to changes in hypothalamic temperature. Measurable changes in cutaneous blood flow result from changes in hypothalamic temperature of tenths of a degree Celsius.
Cutaneous vessels are influenced by reflexes involved in both arterial pressure regulation and temperature regulation. When the appropriate cutaneous vascular responses for temperature regulation and pressure regulation are contradictory, as they are, for example, during strenuous exercise, then the temperature regulating influences on cutaneous blood vessels usually prevail.
Summary
Most of the influences on the medullary cardiovascular centers that have been discussed in the preceding sections are summarized in Figure 9–4. This figure is intended first to reemphasize that the arterial baroreceptors normally and continually supply the major input to the medullary centers. The arterial baroreceptor input is shown as inhibitory because an increase in arterial baroreceptor firing rate results in a decrease in sympathetic output. (Decreased sympathetic output should be taken to imply also a simultaneous increase in parasympathetic output that is not shown in this figure.)
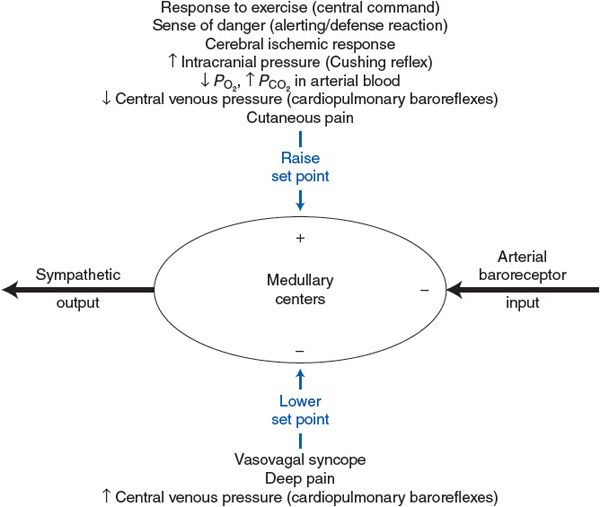
Figure 9–4. Summary of the factors that influence the set point of the arterial baroreceptor reflex.
![]() As indicated in Figure 9–4, the nonarterial baroreceptor influences on the medullary cardiovascular centers fall into two categories: (1) those that increase arterial pressure by raising the set point for the arterial baroreceptor reflex and thus cause an increase in sympathetic activity and (2) those that decrease arterial pressure by lowering the set point for the arterial baroreceptor reflex and thus cause a decrease in sympathetic activity. Note that certain responses that have been discussed are not included in Figure 9–4. The complex combination of stimuli involved in the dive reflex causes simultaneous sympathetic and parasympathetic activation and cannot be simply classified as either pressure raising or pressure lowering. Also, temperature stimuli that discretely affect cutaneous vessels, but not general cardiovascular sympathetic and parasympathetic activity, have not been included in Figure 9–4.
As indicated in Figure 9–4, the nonarterial baroreceptor influences on the medullary cardiovascular centers fall into two categories: (1) those that increase arterial pressure by raising the set point for the arterial baroreceptor reflex and thus cause an increase in sympathetic activity and (2) those that decrease arterial pressure by lowering the set point for the arterial baroreceptor reflex and thus cause a decrease in sympathetic activity. Note that certain responses that have been discussed are not included in Figure 9–4. The complex combination of stimuli involved in the dive reflex causes simultaneous sympathetic and parasympathetic activation and cannot be simply classified as either pressure raising or pressure lowering. Also, temperature stimuli that discretely affect cutaneous vessels, but not general cardiovascular sympathetic and parasympathetic activity, have not been included in Figure 9–4.
The nonarterial baroreceptor influences shown in Figure 9–4 may be viewed as disturbances in the cardiovascular system that act on the medullary cardiovascular centers as opposed to disturbances that act on the heart and vessels. These disturbances cause sympathetic activity and arterial pressure to change in the same direction. Recall from the discussion of the arterial baroreceptor reflex that cardiovascular disturbances that act on the heart or vessels (such as blood loss or heart failure) produce reciprocal changes in arterial pressure and sympathetic activity. These facts are often useful in the clinical diagnoses of blood pressure abnormalities. For example, patients commonly present in the physician’s office with high blood pressure in combination with elevated heart rate (implying elevated sympathetic activity). These same-direction changes in arterial pressure and sympathetic activity suggest that the problem lies not in the periphery but rather with an abnormal pressure-raising input to the medullary cardiovascular centers. The physician should immediately think of those set point–raising influences listed in the top half of Figure 9–4 that would simultaneously raise sympathetic activity and arterial pressure. Often, such a patient does not have chronic hypertension but rather is just experiencing a temporary blood pressure elevation due to the anxiety of undergoing a physical examination.
A more rigorous analysis of the operation of the arterial baroreflex is presented in Appendix E. It is not essential reading, but the serious students may find it enlightening.
LONG-TERM REGULATION OF ARTERIAL PRESSURE
Long-term regulation of arterial pressure is a topic of extreme clinical relevance because of the prevalence of hypertension (sustained excessive arterial blood pressure) in our society. The most long-standing and generally accepted theory of long-term pressure regulation is that it crucially involves the kidneys, their sodium handling, and ultimately the regulation of blood volume. This theory is sometimes referred to as the “fluid balance” model of long-term arterial blood pressure control. In essence, this theory asserts that in the long term, mean arterial pressure is whatever it needs to be to maintain an appropriate blood volume through arterial pressure’s direct effects on renal function.
Fluid Balance and Arterial Pressure
Several key factors in the long-term regulation of arterial blood pressure have already been considered. First is the fact that the baroreceptor reflex, however well it counteracts temporary disturbances in arterial pressure, cannot effectively regulate arterial pressure in the long term for the simple reason that the baroreceptor firing rate adapts to prolonged changes in arterial pressure. (In fact, baroreceptor adaptation may be a good thing. Recall that the whole purpose of arterial pressure is to cause blood to flow through tissues. Thus, it makes little long-term sense to increase arterial pressure by throttling blood flow with reflex arteriolar constriction.)
The second pertinent fact is that circulating blood volume can influence arterial pressure because:
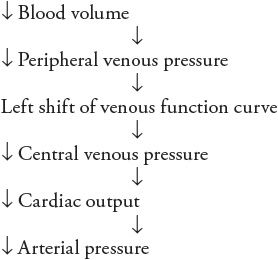
![]() A fact yet to be considered is that arterial pressure has a profound influence on urinary output rate and thus affects total body fluid volume. Because blood volume is one of the components of the total body fluid, blood volume alterations accompany changes in total body fluid volume. The mechanisms are such that an increase in arterial pressure causes an increase in urinary output rate and thus a decrease in blood volume. But, as outlined in the preceding sequence, decreased blood volume tends to lower arterial pressure. Thus, the complete sequence of events that are initiated by an increase in arterial pressure can be listed as follows:
A fact yet to be considered is that arterial pressure has a profound influence on urinary output rate and thus affects total body fluid volume. Because blood volume is one of the components of the total body fluid, blood volume alterations accompany changes in total body fluid volume. The mechanisms are such that an increase in arterial pressure causes an increase in urinary output rate and thus a decrease in blood volume. But, as outlined in the preceding sequence, decreased blood volume tends to lower arterial pressure. Thus, the complete sequence of events that are initiated by an increase in arterial pressure can be listed as follows:
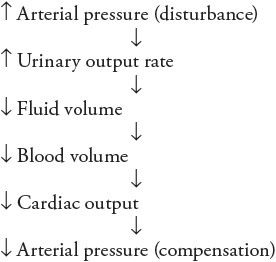
Note the negative feedback nature of this sequence of events: increased arterial pressure leads to fluid volume depletion, which tends to lower arterial pressure. Conversely, an initial disturbance of decreased arterial pressure would lead to fluid volume expansion, which would tend to increase arterial pressure. Because of negative feedback, these events constitute a fluid volume mechanism for regulating arterial pressure.
As indicated in Figure 9–5, both the arterial baroreceptor reflex and this fluid volume mechanism are negative feedback loops that regulate arterial pressure. Although the arterial baroreceptor reflex is very quick to counteract disturbances in arterial pressure, hours or even days may be required before a change in urinary output rate produces a significant accumulation or loss of total body fluid volume. Whatever this fluid volume mechanism lacks in speed, however, it more than makes up for that in persistence. As long as there is any inequality between the fluid intake rate and the urinary output rate, fluid volume is changing and this fluid volume mechanism has not completed its adjustment of arterial pressure. The fluid volume mechanism is in equilibrium only when the urinary output rate exactly equals the fluid intake rate.3 In the long term, the arterial pressure can only be that which makes the urinary output rate equal to the fluid intake rate.4
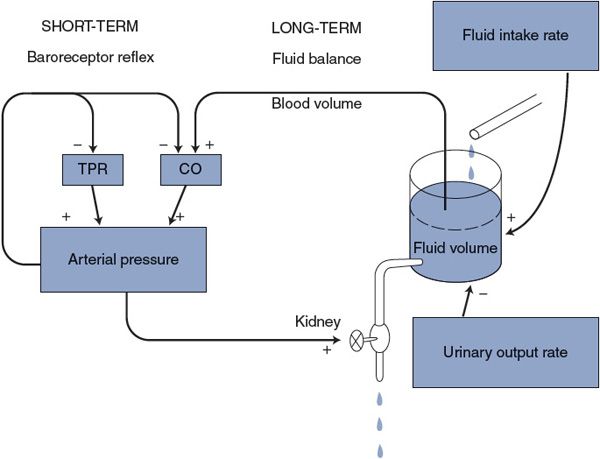
Figure 9–5. Mechanisms of short- and long-term regulations of arterial pressure. TPR, total peripheral resistance; CO, cardiac output.
The baroreceptor reflex is, of course, essential for counteracting rapid changes in arterial pressure. The fluid volume mechanism, however, determines the long-term level of arterial pressure because it slowly overwhelms all other influences. Through adaptation, the baroreceptor mechanism adjusts itself so that it operates to prevent acute changes in blood pressure from the prevailing long-term level as determined through fluid balance.
Effect of Arterial Pressure on Urinary Output Rate
A key element in the fluid balance mechanism of long-term arterial pressure regulation is the effect that arterial pressure has on the renal urine production rate. The mechanisms responsible for this are briefly described here with emphasis on their cardiovascular implications.
The kidneys play a major role in homeostasis by regulating the electrolyte composition of the plasma and thus the entire internal environment. One of the major plasma electrolytes regulated by the kidneys is the sodium ion. To regulate the plasma electrolyte composition, a large fraction of the plasma fluid that flows into the kidneys is filtered across the glomerular capillaries so that it enters the renal tubules. The fluid that passes from the blood into the renal tubules is called the glomerular filtrate, and the rate at which this process occurs is called the glomerular filtration rate. Glomerular filtration is a transcapillary fluid movement whose rate is influenced by hydrostatic and oncotic pressures, as indicated in Chapter 6. The primary cause of continual glomerular filtration is that glomerular capillary hydrostatic pressure is normally very high (![]() 60 mm Hg). The glomerular filtration rate is decreased by factors that decrease glomerular capillary pressure, for example, decreased arterial blood pressure or vasoconstriction of preglomerular renal arterioles.
60 mm Hg). The glomerular filtration rate is decreased by factors that decrease glomerular capillary pressure, for example, decreased arterial blood pressure or vasoconstriction of preglomerular renal arterioles.
Once fluid is filtered into the renal tubules, it either (1) is reabsorbed and reenters the cardiovascular system or (2) is passed along renal tubules and eventually excreted as urine. Thus, urine production is the net result of glomerular filtration and renal tubular fluid reabsorption:
Urinary output rate = Glomerular filtration rate – Renal fluid reabsorption rate
Actually, most of the reabsorption of fluid that has entered renal tubules as glomerular filtrate occurs because sodium is actively pumped out of the renal tubules by cells in the tubular wall. When sodium leaves the tubules, osmotic forces are produced that cause water to leave with it. Thus, any factor that promotes renal tubular sodium reabsorption (sodium retention) tends to increase the renal fluid reabsorption rate and consequently decrease the urinary output rate. The blood concentration of the hormone aldosterone, produced by the adrenal glands, is the primary regulator of rate of sodium reabsorption by renal tubular cells. Adrenal release of aldosterone is, in turn, regulated largely by the circulating level of another hormone, angiotensin II, whose plasma concentration is determined by the plasma concentration of renin, an enzyme that is produced in the kidneys. Renin actually catalyzes the formation of an inactive decapeptide, angiotensin I, from angiotensinogen, a circulating precursor protein produced by the liver. Angiotensin I then gets quickly converted to angiotensin II (an octapeptide) by the action of angiotensin-converting enzyme that is located on the surface of endothelial cells. The combination of elements involved in this whole sequence of events is referred to as the renin–angiotensin–aldosterone system.
The rate of renin release by the kidneys is influenced by several factors. An increase in the activity of renal sympathetic nerves causes a direct release of renin through a β1-adrenergic mechanism. Also, renin release is triggered by factors associated with a lowered glomerular filtration rate. The activation of sympathetic vasoconstrictor nerves to renal arterioles thus indirectly causes renin release via lowered glomerular capillary hydrostatic pressure and glomerular filtration rate. The important fact to keep in mind, from a cardiovascular standpoint, is that anything that causes renin release causes a decrease in urinary output rate because increased renin causes increased sodium (and therefore fluid) reabsorption from renal tubules.5
In the preceding text, we have emphasized the role of the kidneys in regulating blood volume because of its obvious cardiovascular consequences. But regulating bodily fluid volume is only one of the multiple tasks that the kidneys perform. From a renal perspective, a more immediate concern is the regulation of extracellular osmolarity. In fact, osmolarity is one of the most tightly regulated variables in the internal environment. For example, the ingestion of 8 oz (230 mL) of water will elicit a dramatic renal compensatory response within minutes. That is quite amazing when one considers that adding 230 mL of pure water to 42 L of total body water would only reduce internal osmolarity (ie, solute concentration) by approximately ½%.
The kidneys regulate extracellular osmolarity primarily by changing the amount of water (not solutes) in the body. They can do this because they have independent mechanisms for controlling their water and solute excretion rates. Renal water excretion rate is controlled by the hormone vasopressin (also known as antidiuretic hormone) that is released from the posterior pituitary gland. The primary stimulus for vasopressin release is an increase in internal osmolarity, as detected by osmoreceptors in the hypothalamus. Inhibition of vasopressin release occurs with increased afferent input from the cardiopulmonary baroreceptors. Vasopressin increases the water permeability of distal portions of the renal tubules and thus enhances renal water reabsorption and decreases renal water excretion. This is a very rapid and powerful mechanism that essentially keeps internal osmolarity constant in the long term. The bottom line for us is that, given constant internal osmolarity, total extracellular volume must always parallel any changes in total extracellular solute content. Recall that NaCl is by far the most abundant extracellular solute. That is the primary reason that renal sodium handling is of paramount importance in regulating extracellular volume.
Some major mechanisms that influence urinary output rate are summarized in Figure 9–6 with the example of the response to a decrease in arterial pressure. Most important, this figure shows that urinary output rate is linked to arterial pressure by many synergistic pathways. Because of this, modest changes in arterial pressure are associated with large changes in urinary output rate.
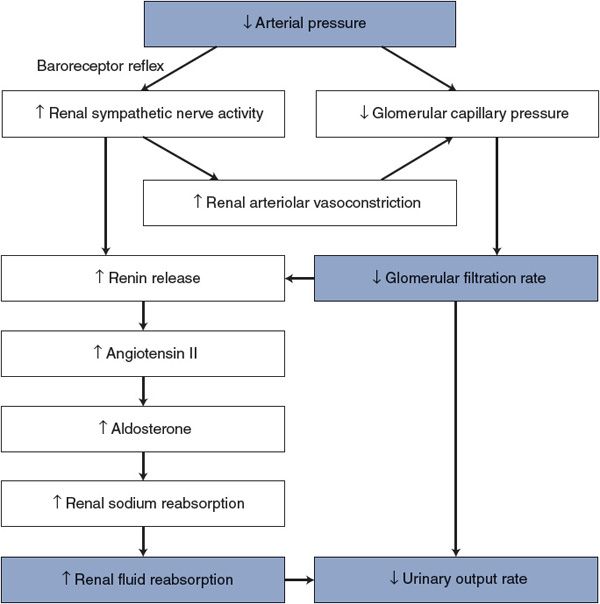
Figure 9–6. Mechanisms by which arterial pressure influences urinary output rate.
The observed relationship between arterial pressure and urinary output for a healthy person is shown in Figure 9–7. Recall that, in the steady state, the urinary output rate must always equal the fluid intake rate and that changes in fluid volume will automatically adjust arterial pressure until this is so. Thus, a healthy person with a normal fluid intake rate will have, as a long-term average, the arterial pressure associated with point A in Figure 9–7. Because of the steepness of the curve shown in Figure 9–7, even rather marked changes in fluid intake rate have minor influences on the arterial pressure of a healthy individual.
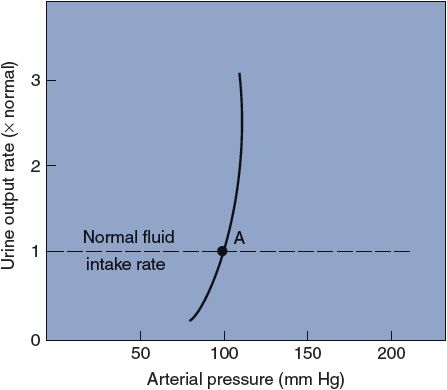
Figure 9–7. The effect of arterial pressure on urinary output rate in a healthy person.
PERSPECTIVES
As will be addressed further in Chapter 11, hypertension (persistent, excessively high arterial pressure) is a common, serious health issue that all physicians will routinely encounter. Almost always, the primary cause is not evident. So, the standard clinical approach is to try to treat the symptom (high arterial pressure) with drugs logically aimed at decreasing either CO or TPR. An ongoing puzzle is that certain drugs that are effective in some patients are not effective in others. Thus, hypertension treatment often proceeds largely on a trial-and-error basis.
In this text, we have emphasized the role of the kidneys and blood volume control in the long-term regulation of arterial pressure for several reasons. At the core of this hypothesis is the undeniable fact that in the long term (months, years, lifetimes) our urinary fluid output must exactly match our highly variable fluid input. Otherwise we would gradually either desiccate to cinders or turn into huge water-filled blobs. Any hypothesis about long-term control of arterial pressure (or any other life variable for that matter) must ultimately work within this constraint. Moreover, the two known (but admittedly very rare) definite causes of hypertension (renal artery obstruction or excess aldosterone production from an adrenal gland adenoma) both point to the kidneys.
However, in view of the variable clinical experience in dealing with hypertension, it is well to question whether the kidneys are the sole organs responsible for long-term arterial pressure regulation. Indeed, many argue that the CNS is intimately involved as well. One common hypothesis along these lines is that: “Long-term arterial pressure is regulated to be whatever it needs to be to maintain adequate brain blood flow.” This obviously is somewhat a different mindset than: “Long-term arterial pressure is regulated to be whatever it needs to be to make urine output rate equal to fluid intake rate.” Certainly, the very existence of the “Cushing reflex” lends some credence to the CNS hypothesis. Moreover, even the short-term regulation of arterial pressure is a complex issue that can involve many inputs to the cardiovascular control centers in addition to those from the arterial baroreceptors (see Figure 9–4). It is largely unknown to what extent these “other” influences might be involved in long-term pressure regulation. The bottom line of all this is that the interplay of all the factors involved in the long-term control of arterial pressure is still an active topic of debate.
We remind the student that, whatever the purpose of long-term arterial pressure regulation, changes in mean arterial pressure can be accomplished only by changing cardiac output and/or total peripheral resistance. Blood volume is undeniably one important determinant of cardiac output. Thus, we (the authors of this text) conclude that there are elements of truth in both the renal and CNS theories of long-term blood pressure control. Hopefully, this debate will ultimately result in some melding of the theories that will improve our understanding of what factors cause chronic hypertension.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


