Chapter 44 Radionuclide Imaging of Inflammation in Atheroma
INTRODUCTION
Over 16 million people in the United States1 are afflicted with coronary artery disease. Approximately 785,000 of these individuals will experience a sudden coronary event in 2009, and about 152,000 will succumb to the acute process. Although identification of each person at risk is the ultimate clinical goal, it is most desirable to definitively identify the subset of patients who are most likely to suffer an acute coronary event. In 60% to 80% of patients, the acute event is caused by thrombotic occlusion of the vessel due to rupture of an atheroma in the coronary artery; in a minority (especially patients < 50 years old and smokers), the occlusion results from erosion of endothelial cells overlying the plaque (likely due to apoptosis of endothelial cells, exposing thrombogenic proteoglycans and microparticulates2). Lesions undergoing plaque rupture occupy a significant fraction of the circumference of the vessel, contain numerous inflammatory cells in the necrotic core, have a thin cap (<65 μm) separating the atheroma from blood in the vessel lumen, and have a marked increase in vasa vasorum.3 Diagnostic targeting strategies should identify plaques vulnerable to rupture or endothelial erosion.
EVOLUTION OF ATHEROSCLEROTIC LESIONS
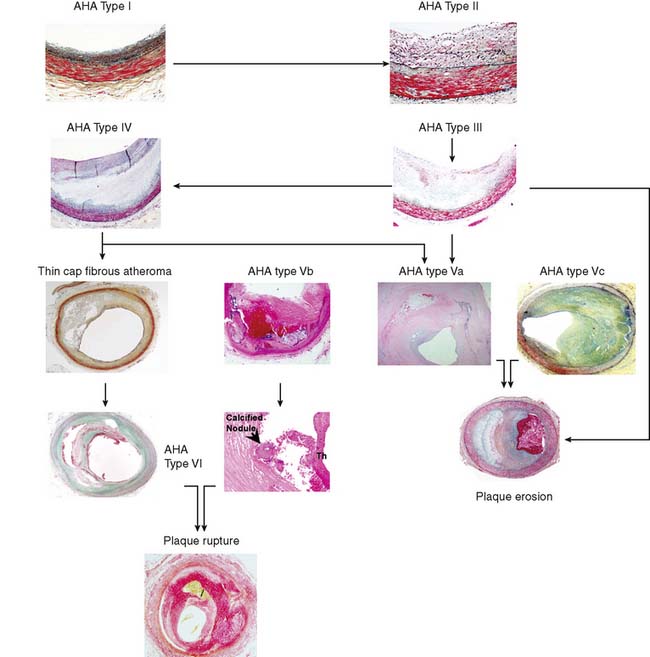
The American Heart Association (AHA) classifies atherosclerotic lesions from type I through VI based on the severity of the disease (Fig. 44-1).4 Type I plaque is an adaptive lesion consisting mostly of smooth muscle cells (SMC) with or without scattered macrophage infiltration. Type II plaque is a fatty streak consisting of foam cell infiltration. Type III plaque shows pools of extracellular lipid, but no necrotic core is formed; the extracellular matrix predominantly consists of proteoglycans. Type IV plaque has a well-defined necrotic core with overlying fibrous cap consisting of SMC and evolving collagenous matrix. Type V lesions demonstrate a thick fibrous cap consisting of a predominantly fibrotic lesion (type Vc) with calcification (type Vb) or deep-seated necrotic core (type Va). Type VI is a classic plaque rupture that shows a region of fibrous cap disruption that allows continuation between the necrotic core and overlying luminal thrombus. Although this histologic classification has neat groupings, clinically, patients do not progress linearly from types I to VI. To make the classification more relevant, several authors have proposed modifications to this classification system. One such classification suggested by Virmani and colleagues5 classifies lesions based on the morphologic description of the fibrous cap and neointima and their progression to complicated lesions. They have categorized atherosclerotic plaques as “intimal thickening” (equivalent to AHA type I), “intimal xanthoma” (type II), “pathologic intimal thickening” (type III), and “fibrous cap atheroma” (similar to types IV and V). The unique features of the Virmani classification include linking of pathologic intimal thickening (SMC + proteoglycan-rich lesions, equivalent to AHA type III) to acute coronary syndromes associated with plaque erosion, and description of an entity referred to as thin fibrous cap atheroma. The latter is a precursor of plaque rupture–related acute coronary syndromes. This classification has incorporated repeated cycles of healing on plaque ruptures and erosions toward development of more advanced lesions. On the other hand, the newer classification also assigns significance to calcific nodules in predisposition to the luminal thrombus. The morphologic characteristics of the atherosclerotic lesions described in the new classification are compared with the AHA classification in Figure 44-1.
PATHOGENETIC BASIS OF INFLAMMATION IN ATHEROSCLEROSIS
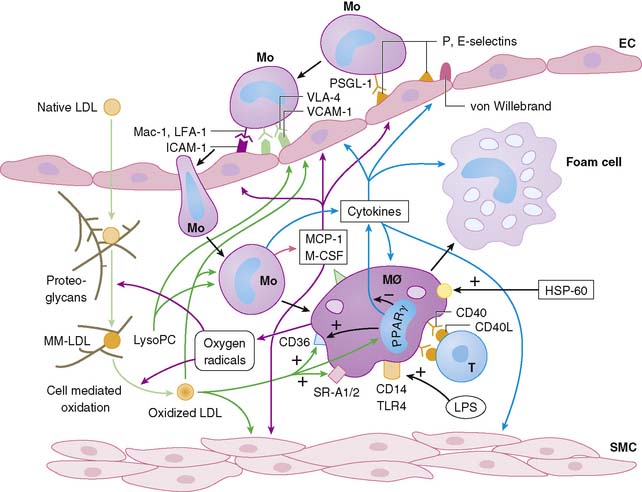
Atherosclerosis involves both an inflammatory response to lipid deposition in the vessel wall and an immunologic response of the endothelium to the injury. Damage to the vascular intima is initiated by various factors, including shear stress and, most important, hyperlipidemia.6–8 Endothelial damage causes expression of selectins and adhesion molecules by the endothelial cell. These chemotactic factors cause recruitment of circulating monocytes to the region of injury; these cells migrate into the subendothelial layer. There, the short-lived monocytes are transformed into long-lived macrophages.9 The macrophages phagocytize and start to metabolize low-density lipoprotein (LDL) cholesterol. In the process of metabolism, oxidative products are generated, leading to the formation of oxidized LDL cholesterol, both in the intracellular and extracellular environment. Oxidized LDL cholesterol causes greater inflammation, is difficult to metabolize, and in high concentration is toxic to the macrophages. Modified (mainly oxidized) LDL enters the macrophages through scavenger LDL receptors, which are not inhibited by the intracellular lipid contents, allowing the cells to continue gorging themselves on this relatively indigestible irritant. As large quantities of LDL cholesterol and oxidized LDL cholesterol accumulate in the macrophages, they become lipid-laden foam cells. Large quantities of intracellular oxidized LDL cholesterol causes death of the macrophages, in part by apoptosis10 and in part due to necrosis. Part of the process of apoptosis is production of caspase-1 by the mitochondria of the macrophage. Caspase-1 production is associated with increased production of matrix metalloproteinases, resulting in degradation of stromal tissue containing the lesion.11 These foam cells are restricted from moving away from the subintimal space. Concurrent with the release of selectins and attraction of monocytes to the site of injury, the injured endothelium releases substances that induce phenotypic alteration of medial SMC from contractile cells to the proliferating phenotype. The transformed smooth muscle cells migrate to the neointima.
The macrophage infiltration in the vessel wall follows a systematic process that includes reversible adhesion of the monocytes to the injured endothelium, followed by monocytic activation, leading to a more permanent binding and eventual subendothelial migration of monocytes (Fig. 44-2).12 The initial injury to endothelial cells induces expression of integrin molecules such as E-selectin or P-selectin. Corresponding integrin moieties on the monocytes, such as L-selectin, facilitate interdigitating interaction between the endothelium and circulating monocytes that slows the monocytes as they start to roll along the vessel wall. Chemotactic peptides cause the monocytes to adhere to the injured endothelium. In the absence of chemoattractants, the selectins are shed, and the monocytes roll back to the bloodstream. The interaction of the endothelial chemoattractants and their receptors on monocytes lead to activation of β1– or β2-integrins (such as LFA-1, VLA-4, or Mac-1). These integrins bind firmly to endothelial expression of adhesion molecules of an immunoglobulin gene superfamily such as intercellular adhesion molecule 1 (ICAM-1), ICAM-2, and VCAM-1. The multipronged attachment of the monocytes to the endothelium commits monocytes to permeate through the endothelial cell junctions, mediated by co-adherins. In addition to the monocyte/macrophage infiltration, both PMNs and mast cells appear to play important roles in plaque rupture. Immune histology studies demonstrated that mast cells localize in the shoulders and cap of the plaque, suggesting a role for these cathepsin G–producing cells in plaque rupture.13
APPROACHES TO IMAGING ATHEROMA
There have been multiple radionuclide approaches to imaging atheroma: