Pulmonary Surfactant and Disorders of Surfactant Homeostasis
INTRODUCTION
Pulmonary surfactant is a complex mixture of phospholipids and proteins that creates a unique interface separating alveolar gas and liquids at the alveolar cell surface, reducing surface tension, and maintaining lung volumes at end expiration. Reduction of the surface tension at the air–liquid interface is a requirement for respiratory function following birth. Deficiency of pulmonary surfactant causes respiratory failure in premature infants, or infantile respiratory distress syndrome (IRDS). The adequacy of pulmonary surfactant is maintained by unique and highly regulated systems mediating the synthesis, secretion, reutilization, and catabolism of surfactant. Loss or inactivation of pulmonary surfactant later in life occurs in the adult respiratory distress syndrome (ARDS), a significant cause of morbidity and mortality following infection, shock, or trauma. Mutations in genes regulating surfactant homeostasis, including SFTPA, SFTPB, SFTPC, ABCA3, TITF1, and CSF2RA cause acute and/or chronic lung disease in newborn infants, children, and adults. Disorders of GM-CSF signaling inhibit surfactant lipid and protein catabolism by alveolar macrophage causing pulmonary alveolar proteinosis (PAP). This chapter reviews the biology of the surfactant system and its implications for the pathogenesis, diagnosis, and treatment of respiratory disease in premature infants and adults. Suggested reviews of these topics are provided in the References section.1–5
PHYSICAL FORCES AT THE AIR–LIQUID INTERFACE
In 1929, Van Neergard recognized the critical role of surface tension as a “retractile force” in the lung, observing the marked difference in inflation pressures required to inflate the air- versus water-filled lung. Avery and Mead associated the lack of a lipid-rich material in the lungs of infants dying from idiopathic respiratory distress syndrome with alveolar collapse and respiratory failure.6 In the absence of pulmonary surfactant, molecular forces at the air–liquid interface create a region of high surface tension because intermolecular forces between water molecules are unopposed at the air–liquid interface, and an area of high retractile force at the surface is created. Forces of 70 dynes/cm2 are generated at the air–water interface; if unopposed in the alveolus, such forces lead to alveolar collapse and respiratory failure. A surface film composed of multilayered sheets of phospholipids creates a distinct phase separating air and liquid, reducing surface tension to nearly zero and maintaining residual lung volume at end expiration. Complex interactions between surfactant phospholipids and proteins are required to maintain surfactant film throughout life. Pulmonary surfactant lipids and proteins are synthesized and secreted by alveolar type II epithelial cells into the alveoli, where they form multilayered lipid-rich films that reduce surface tension to maintain ventilation (Figs. 5-1 and 5-2).
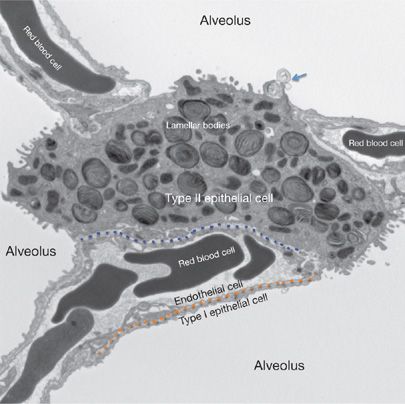
Figure 5-1 Pulmonary alveolar ultrastructure. The air–blood barrier is comprised of the capillary endothelium (above the dotted orange line) and the closely apposed type I epithelial cell (below the dotted orange line). The dotted blue line delineates the interstitial space between endothelial cells and type II epithelial cells with their specialized secretory lipid organelles (lamellar bodies). Type II cells form tight junctions with type I cells and serve as alveolar progenitor cells. Surfactant lipids and proteins are secreted as lamellar membranes (arrow) into the alveolar space forming tubular myelin and lipid membrane multilayers that reduce surface tension, preventing alveolar collapse.
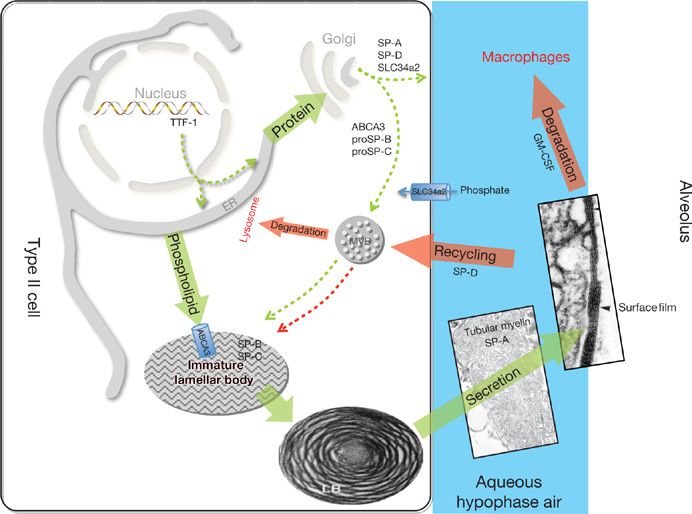
Figure 5-2 Surfactant metabolism. Newly translated surfactant proteins (proSP-B and proSP-C) and lamellar body (ABCA3) proteins traffic from the endoplasmic reticulum (ER) to the Golgi and subsequently to the multivesicular body (MVB). Fusion of the MVB with the lamellar body (LB) is accompanied by proteolytic processing of SP-B and SP-C proproteins to their mature peptides. Surfactant phospholipids (DPPC, PG) are likely transported directly from the ER to the LB by lipid transfer proteins. The contents of the LB are secreted into the alveolar space where they interact with SP-A to form tubular myelin and, ultimately, a phospholipid-rich film (surfactant) at the air–liquid interface. Alveolar surfactant lipids and proteins are cleared through a GM-CSF dependent pathway that regulates alveolar macrophage differentiation and function. Surfactant remnants are also taken up by the type II epithelial cell and recycled to the LB, via the MVB, for resecretion, while a portion is degraded in lysosomes. SP-D plays an important role in regulating alveolar surfactant pool size likely by enhancing its reuptake by type II epithelial cells. The MVB serves to integrate surfactant synthesis, secretion, recycling, and degradation pathways in the type II cell. TTF-1 is a transcription factor critical for differentiation of type II epithelial cells and regulation of expression of Abca3, Slc34a2, and the surfactant proteins. Synthetic pathways are shown in green and catabolic pathways are shown in red.
COMPOSITION OF PULMONARY SURFACTANT
Pulmonary surfactant isolated by lung lavage consists of highly heterogeneous forms of phospholipid–protein aggregates of distinct sizes, structural characteristics, and composition. Tubular myelin is the most abundant form of alveolar phospholipid and consists of large, relatively dense aggregates (termed large aggregate surfactant) composed of phospholipids and surfactant proteins (SPs). Tubular myelin is a highly organized form of surfactant phospholipid, forming square tubular arrays. Tubular myelin represents an extracellular pool of surfactant lipids that rapidly moves to the air–liquid interface and reorganizes to form multilayered sheets that reduce surface tension in the alveolus (Fig. 5-2). Large lamellated structures, with lipid composition similar to that of tubular myelin, are seen within the alveolus and likely represent newly secreted lamellar bodies that unravel to form tubular myelin in the alveoli. The phospholipid composition of lamellar bodies, the intracellular storage form of surfactant, tubular myelin, and lamellated forms present in the alveolus are virtually identical. Smaller, less dense particles (small aggregate surfactant) are also present within the alveolar space, representing remnants or catabolic forms of surfactant that have relatively poor surface activity. Small aggregate surfactant is destined for uptake, reutilization, or catabolism by type II epithelial cells and catabolism by alveolar macrophages.2,3
 SURFACTANT PHOSPHOLIPIDS AND PROTEINS
SURFACTANT PHOSPHOLIPIDS AND PROTEINS
The composition of surfactant lipids is similar in all of the structural forms of surfactant isolated from mammalian lungs, with phospholipids generally representing 80% to 90% of the mass of pulmonary surfactant.3 In the adult lung, phosphatidylcholine (PC) and phosphatidylglycerol (PG) are the most abundant phospholipids, representing approximately 70% to 80% and 5% to 10%, respectively, of the lipid mass. Dipalmitoyl phosphatidylcholine (DPPC) is the most abundant species of PC. Lesser amounts of phosphatidylserine, phosphatidylethanolamine, sphingomyelin, neutral lipids (mostly cholesterol), and glycolipids are also present in surfactant. The lung content of surfactant phospholipids increases markedly with advancing gestation, regulated by a complex signaling and transcriptional network that controls type II alveolar cell differentiation, lipid synthesis, and SP gene expression. Lamellar bodies are secreted into the fetal amniotic fluid. PC, lamellar body counts, DPPC content, and increased lecithin (PC) to sphingomyelin (L/S) ratio, correlate with postnatal respiratory function. These tests are used to predict pulmonary maturity prior to the birth of preterm infants. Lung maturation and synthesis of surfactant components are induced by maternal administration of glucocorticoids, used clinically to prevent respiratory distress prior to premature birth.7 Proteins represent approximately 5% to 15% of the mass of pulmonary surfactant and include serum proteins and proteins that are synthesized and secreted by type II alveolar epithelial cells. In addition to its specific interaction with SP-B, the anionic phospholipid PG may also play an important role in innate defense. PG constitutes 10 mole% of surfactant lipid, with palmitoyl-oleoyl-PG (PoPG) being the most common species in human surfactant. PoPG specifically suppresses LPS-induced inflammatory responses and prevents infection of epithelial cells by binding RSV or influenza A virus.8 Thus the unique enrichment of PoPG in the distal airspaces may be an important component of host defense against inhaled pathogens. Four surfactant proteins (SPs), SP-A, SP-B, SP-C, and SP-D, are produced by respiratory epithelial cells, each playing specific roles in surfactant homeostasis and innate host defense.2,3,9,10
Surfactant is uniquely enriched in disaturated DPPC. The saturated C16 acyl chains pack densely at an air–liquid interface, reducing tension at the surface. However, dense and stable packing of DPPC occurs at a phase transition of 41°C, far above physiologic temperatures. Thus, at 37°C, pure DPPC maintains a semicrystalline or gel phase that is incapable of moving rapidly with the expansion and compression of the alveoli during the respiratory cycle. The capability of DPPC pulmonary surfactant to move rapidly to the alveolar interface at 37°C and to maintain low surface tension during dynamic compression is conferred by the surfactant-associated proteins SP-B and SP-C. PC synthesis in the lung is controlled by genes encoding choline phosphate cytidylyltransferase (PCYTLA) and choline kinase (CHKA), which are required for surfactant lipid synthesis and lung function at birth. DPPC is synthesized in type II alveolar cells via both a de novo pathway and remodeling of lysoPC. The enzyme lysoPC acetyltransferase (LPCAT1) mediates reacylation during surfactant lipid biosynthesis. Surfactant lipids, synthesized in the endoplasmic reticulum (ER) are transferred via a Golgi-independent pathway to lamellar bodies, the major intracellular storage site of surfactant (Fig. 5-2). Transfer of lipids occurs via nonvesicular transport and uptake into lamellar bodies requires the ABCA3 transporter, which selectively transports PC and PG. In contrast, surfactant proteins SP-B and SP-C traffic from the ER to the Golgi and subsequently to multivesicular bodies where proteolytic processing is initiated. Ultimately, multivesicular bodies are incorporated into lamellar bodies with surfactant lipids prior to secretion from type II alveolar cells.3
 STRUCTURE AND FUNCTION OF SURFACTANT PROTEINS
STRUCTURE AND FUNCTION OF SURFACTANT PROTEINS
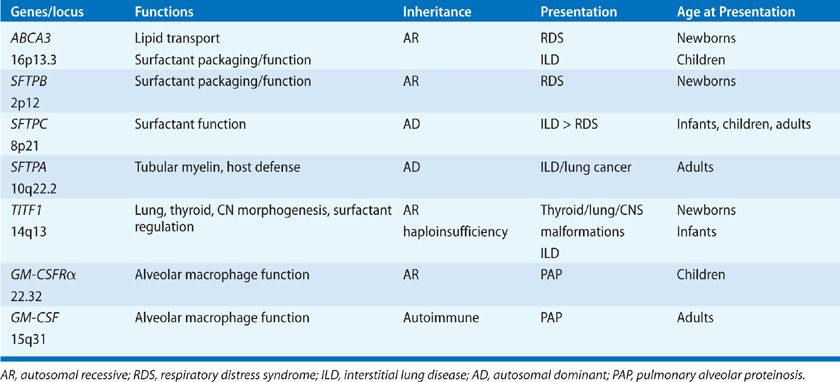
Four distinct surfactant-associated proteins have been isolated from surfactant obtained by lung lavage. Their cDNAs, genes, and structures have been identified and are well characterized (Table 5-1).9,10 The SPs are expressed in a relatively lung epithelial cell–selective manner and are secreted into the airspaces, where they influence the structure, metabolism, and function of surfactant. Two classes of proteins have been distinguished on the basis of their structures. SP-A and SP-D are relatively abundant, hydrophilic, structurally related proteins and are members of the calcium-dependent lectin family of proteins that have similar amino-terminal collagenous and C-terminal lectin domains.11 SP-A and SP-D have little “surfactant”-like qualities but are able to bind complex carbohydrates, lipids, and glycolipids, including those on the surface of cells, bacteria, viruses, fungi, and other lung pathogens. SP-A and SP-D influence the structural forms and metabolism of surfactant lipids in the alveolus. They act as opsonins, activate alveolar macrophages, and play important roles in innate host defense in the lung. In contrast, SP-B and SP-C are small, hydrophobic proteins that play critical roles in enhancing the rate of spreading and stability of surfactant phospholipids needed to optimally reduce surface tension.10 SP-B and SP-C are the sole protein components of the animal-derived surfactant replacement preparations used for the treatment of IRDS at present.10
TABLE 5-1 Regulation of Surfactant Homeostasis
Surfactant Protein B (SP-B)
SP-B is a hydrophobic, amphipathic 8.8-kDa protein produced from a single gene (SFTPB, OMIM 178640) located on human chromosome 2. The SP-B mRNA is expressed in nonciliated bronchiolar cells and type II alveolar cells and is translated to produce a 40- to 42-kDa precursor that is proteolytically processed in the secretory pathway of type II epithelial cells to form the active 79-amino acid peptide found in alveolar surfactant. In combination with lipids, SP-B can reconstitute most of the surface activity of natural lung surfactant. SP-B contains two regions, (Trp9-Pro23) and (Ile56-Pro67), predicted to form amphipathic α-helices that interact with the surface of lipid films. Almost 50% of the protein is in an α-helical conformation; the amphipathic domains of SP-B interact with surfactant lipids, and PG in particular, to promote lipid incorporation into and stabilization of the surface film. SP-B contains three intramolecular disulfide bonds that confine the amphipathic helices of SP-B in an antiparallel configuration. Intermolecular disulfide bonds stabilize SP-B dimers. Dimers and higher multimers of SP-B, which are probably stabilized by noncovalent interactions, are readily identified in pulmonary surfactant.
ProSP-B is synthesized in the ER and proteolytically processed in multivesicular and lamellar bodies by cathepsins and other intracellular proteases. The active SP-B peptide is packaged with SP-C and surfactant lipids in lamellar bodies prior to secretion into the alveolus. In the alveoli, the positively charged amino acid residues of SP-B selectively interact with the negatively charged phospholipid DPPG. In a mixed DPPC/DPPG monolayer, SP-B is believed to purify the DPPC monolayer by removal of DPPG. SP-B increases order in the lipid head groups with little effect on order on acyl chains in the lipid membrane interior. The ability to order the lipid head group region is located in the amino- and carboxy-terminal regions of SP-B (1–20) and (53–78), which contain the predicted amphipathic helices. Synthetic peptides that contain these two regions have surface-tension–lowering activity similar to that of native SP-B and peptide mimics have been developed for therapy of respiratory distress in infants. SP-B enhances the insertion (fusion) of phospholipid vesicles into a preformed DPPC/DPPG monolayer, particularly in the presence of divalent cations. SP-B causes lipids in solution to form discoid particles often appearing as stacks or sheets. Together with SP-A, lipids, and Ca2+, SP-B reconstitutes the characteristic ultrastructural features of tubular myelin, producing multilamellar aggregates and square lattice configurations.
SP-B is Required for Survival After Birth Mutations in the gene encoding SP-B (SFTPB, OMIM 265120) cause acute respiratory failure at birth related to surfactant dysfunction.12,13 Similarly, deletion of Sftpb in the developing lung or its conditional deletion in adult mice causes acute respiratory distress associated with alveolar capillary leak and surfactant deficiency.14 Pathologic findings of patients with lung disease related to SFTPB mutations are similar to those in mice in which the Sftpb gene is deleted.14,15 SP-B deficiency is associated with failure to form lamellar bodies, accumulation of abnormal multivesicular bodies within the type II cells, and failure to form tubular myelin or functional surfactant films after secretion into the alveoli.
Hereditary SP-B Deficiency Causes Respiratory Failure at Birth SP-B deficiency is inherited in an autosomal recessive pattern, affected infants developing respiratory failure shortly after birth. While lung morphogenesis proceeds normally in utero, the lack of SP-B results in acute atelectasis and respiratory distress, usually presenting as full-term infants with signs and symptoms of diffuse alveolar collapse after birth. More than 40 distinct SFTPB mutations in patients with SP-B deficiency have been identified.13,15 The disorder is refractory to surfactant replacement therapy and most patients die from respiratory failure within several months after birth, requiring oxygen and ventilatory support throughout their clinical course. Lung transplantation has been offered to some patients. SP-B deficiency disrupts the formation of lamellar bodies and tubular myelin, and interferes with the processing of proSP-C to the active peptide. Thus, most SP-B–deficient patients lack both SP-B and SP-C peptides in the alveolus. In patients with SFTPB-related disease, proSP-C accumulates in the airspaces, contributing to an alveolar proteinosis–like syndrome. Pathologic diagnoses include desquamative interstitial pneumonitis (DIP), chronic pneumonitis of infancy (CPI) or infantile alveolar proteinosis, histologic changes being influenced by age, and supportive therapies (Fig. 5-3).16 The definitive diagnosis is made by identification of SFTPB gene mutations, enabling prenatal diagnosis and genetic counseling. While various missense, nonsense, frameshift, and splice variants have been identified, 121ins2 mutation in exon 4 is the most common mutation.12 In most affected infants, SP-B is lacking in bronchoalveolar lavage fluid and the abnormal proSP-C peptide accumulates in the alveoli, findings that can be verified by immunohistochemistry. Patients with SP-B deficiency do not respond to surfactant replacement and generally succumb from chronic respiratory failure early in infancy in spite of intensive care.
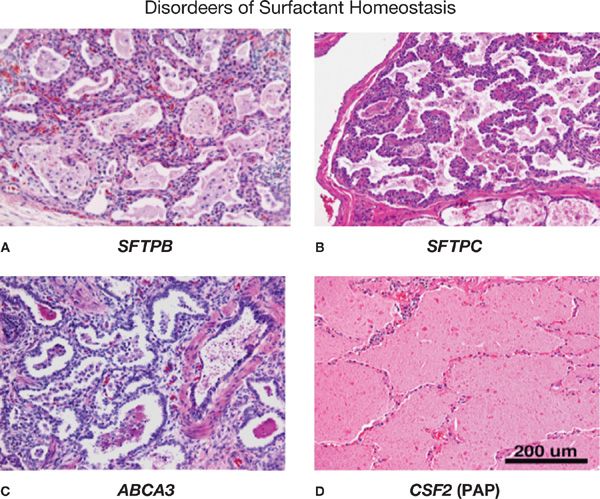
Figure 5-3 Pulmonary histopathology associated with disorders of surfactant homeostasis. Pathologic findings in eonates with mutations in SFTPB (A), SFTPC (B), and ABCA3 (C) are consistent with varying pathologic diagnoses, for example, childhood interstitial pneumonitis (CIP), nonspecific interstitial pneumonitis (NSIP), desquamating interstitial pneumonitis (DIP) or pulmonary alveolar proteinosis (PAP) (D). Severe alveolar remodeling, alveolar loss, macrophage infiltration, varying degrees of alveolar proteinosis, and stromal thickening are observed. In contrast, auto-antibodies against GM-CSF or mutations in the GM-CSF receptor (CSFR2A) are associated with pulmonary alveolar proteinosis in which surfactant lipids and proteins accumulate in the alveolus. Alveolar structure is generally well maintained in PAP. (Reproduced with permission from Whitsett JA, Wert SE, Trapnell BC: Genetic disorders influencing lung formation and function at birth. Hum. Mol. Genet. 2004;13:R207–R215.)
Surfactant Protein C (SP-C) In humans, SP-C is encoded by a single gene (SFTPC, OMIM 178620), located on human chromosome 8.2,10 SP-C mRNA is expressed exclusively in type II epithelial cells in the lung and is translated to produce a 22-kDa precursor that is palmitoylated and proteolytically processed during intracellular transport to form the active, hydrophobic peptide of 35 amino acids stored in lamellar bodies.2,3 After secretion, SP-C enhances the surface-active properties of lipid mixtures, lowering surface tension during compression, and enhancing adsorption rate of lipid films at the air–water interface. Both SP-C and SP-B enhance the speed of formation and stability of lipid films. A mixture of surfactant lipids and proteins SP-B and SP-C improves lung inflation and compliance and is useful for treatment of respiratory distress syndrome (RDS) in newborn infants. SP-C is palmitoylated on cysteine residues near the NH2
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


