1. Pulmonary arterial hypertension
1.1 Idiopathic PAH
1.2 Heritable
1.2.1. BMPR2
1.2.2. ALK1, endoglin, SMAD9, CAV1, KCNK3
1.2.3 Unknown
1.3 Drug- and toxin-induced
1.4 Associated with
1.4.1. Connective tissue diseases
1.4.2 HIV infection
1.4.3 Portal hypertension
1.4.4 Congenital heart diseases
1.4.5 Schistosomiasis
1′. Pulmonary veno-occlusive disease (PVOD) and/or pulmonary capillary hemangiomatosis (PCH)
1′′. Persistent pulmonary hypertension of the newborn
2. Pulmonary hypertension due to left heart disease
2.1 Systolic dysfunction
2.2 Diastolic dysfunction
2.3 Valvular disease
3. Pulmonary hypertension due to lung diseases and/or hypoxia
3.1 Chronic obstructive pulmonary disease
3.2 Interstitial lung disease
3.3 Other pulmonary diseases with mixed restrictive and obstructive pattern
3.4 Sleep-disordered breathing
3.5 Alveolar hypoventilation disorders
3.6 Chronic exposure to high altitude
3.7 Developmental abnormalities
4. Chronic thromboembolic pulmonary hypertension (CTEPH)
5. PH with unclear multifactorial mechanisms
5.1 Hematologic disorders: chronic hemolytic anemia, myeloproliferative disorders splenectomy
5.2 Systemic disorders, sarcoidosis, pulmonary Langerhans cell histiocytosis, lymphangioleiomyomatosis, neurofibromatosis, vasculitis
5.3 Metabolic disorders: glycogen storage disease, Gaucher disease, thyroid disorders
5.4 Others: tumoral obstruction, fibrosing mediastinitis, chronic renal failure
Classification of Pulmonary Hypertension
The classification of PH adopted in the ERS/ESC guidelines and recently revised during the 5th Word symposium on PH is presented in Table 33.1 [1, 3]. The group 1 corresponds to all forms of PAH, including idiopathic PAH, heritable PAH, drugs and toxins induced PAH, and PAH associated with different conditions (connective tissue diseases, HIV infection, portal hypertension, congenital heart disease, schistosomiasis). A sub-group 1′ includes a rare entity characterized by a predominant pulmonary venous or capillary involvement: pulmonary veno-occlusive disease (PVOD) and pulmonary capillary hemangiomatosis (PCH). Pathologic and genetic studies have recently demonstrated that PVOD and PCH represents distinct naming of the same entity [4–8]. Group 2 includes post-capillary PH due to left heart diseases, defined by an increased pulmonary capillary wedge pressure (above or equal to 15 mmHg). Group 3 was defined as “PH due to chronic lung diseases and/or hypoxia”. In this group, the predominant cause of PH is hypoxemia as a result of either chronic lung disease, impaired control of breathing, or residence at high altitude; however, the precise prevalence of PH in all these conditions remains unknown. In this group, combined pulmonary fibrosis and emphysema represents a category of lung disease characterized by a mixed obstructive and restrictive pattern frequently associated with severe PH [9]. Group 4 defined chronic thromboembolic pulmonary hypertension. Group 5 corresponds to heterogeneous conditions with unclear or multifactorial etiologies. This group includes notably sarcoidosis, pulmonary Langerhans’ cell histiocytosis, lymphangioleiomyomatosis, and neurofibromatosis type 1.
In the evaluation of PH occuring in the context of orphan lung diseases, physicians should rule out other types of PH (in particular post-capillary PH and CTEPH) and screen for risk factors of PAH (connective tissue diseases, portal hypertension, HIV infection…). Of note, precapillary PH associated with orphan lung diseases may be observed in different subgroups of this classification: in group 3 (syndrome of combined pulmonary fibrosis/emphysema), group 5 (sarcoidosis, pulmonary Langerhans cell histiocytosis, lymphangioleiomyomatosis, neurofibromatosis) and group 1′ (PVOD/PCH).
Pulmonary Hypertension Associated with Sarcoidosis
Sarcoidosis is a multisystem disease characterized by granulomatous inflammation of unknown cause, with pulmonary involvement being one of the commonest disease manifestations. PH may complicate sarcoidosis with an estimated prevalence of 1–28 % [10–13]. Pathological processes underlying sarcoidosis-associated PH (SAPH) are complex and multiple [10], and may fall under different groups according to the current clinical classification of PH [3]. A true vasculopathy with arterial or venous infiltration by granulomas may occur, generating venular lesions mimicking PVOD (Fig. 33.1) [14–16]. Cardiac sarcoidosis may contribute to post-capillary PH, mainly through diastolic dysfunction (myocardial involvement) [17]. Parenchymal lung disease related to sarcoidosis can result in extensive interstitial fibrosis with destruction of the pulmonary vascular bed, and together with alveolar hypoxia, may promote the development of mild or moderate precapillary PH [10, 13, 17]. Enlargement of intrathoracic lymph nodes or fibrosing mediastinitis can lead to extrinsic compression of the proximal pulmonary vasculature [14, 18, 19]. Furthermore, hepatic involvement may exceptionally result in porto-pulmonary hypertension [20]. The often multifactorial nature of SAPH is best considered under a specific subgroup in the classification of PH – “unclear and/or multifactorial etiology” [3] (Table 33.1).
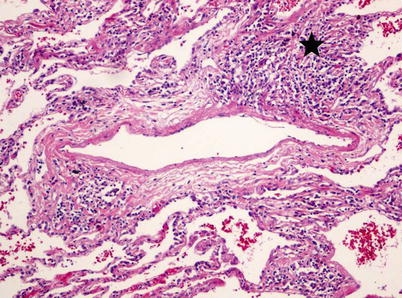
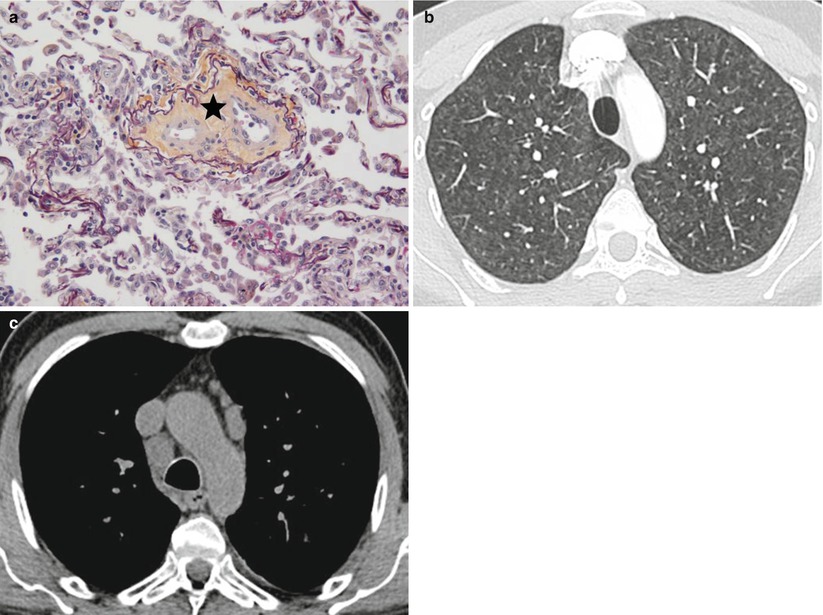
Fig. 33.1
Pathologic assessment of a patient with pulmonary hypertension associated with sarcoidosis. Pulmonary venous involvement is frequently observed in pulmonary hypertension associated with sarcoidosis. Epitheloid giant-cell granulomas can be observed in the vicinity of veins, and may lead to their obstruction. Magnification 100, hematoxylin-eosin staining
Although PH is frequently associated with advanced fibrotic lung disease in sarcoidosis [10, 15, 21], there is occasionally a significant discrepancy between the severity of lung disease with the severity of PH. This suggests that alternate mechanisms, other than direct obliteration of the vascular bed by the fibrotic process, may participate to the development of PH [10, 14]. In absence of parenchymal involvement, distal granulomatous vasculopathy should be suspected [13–17, 22, 23]. Notably, pulmonary venular involvement have been frequently reported [14].
Prevalence of PH in sarcoidosis patients vary largely according to the population studied. It has been estimated to be around 5–15 % in unselected patients with sarcoidoisis, up to 50 % in patients with persistent dyspnea [8, 14, 16, 22, 24] and as high as 75 % in patients with advanced parenchymal lung disease on transplantation waiting list [12]. Nevertheless, interpretation of the true prevalence of PH in sarcoidosis is limited because right heart catheterization was not performed in many studies, despite the low accuracy of echocardiography to detect PH and confirm its mechanism [17]. Several studies have demonstrated a correlation between severity of the disease and PH occurrence, in particular for mild or moderate PH [10, 12, 21]. Moreover, PH in sarcoidosis has been associated with oxygen desaturation during 6 min walking test and low DLCO [17]. However, PH may be present in all stages of sarcoidosis [10, 14] and referral for formal RHC is mandatory if PH is suspected [17]. Interestingly, others biomarkers of PH such as NT-proBNP have failed to predict PH occurrence in sarcoidosis, although it has been demonstrated to be increased in cardiac involvement [25].
It is recognized that patients with SAPH have a worse prognosis compared to those without pre-capillary PH [21]. Five-year survival in SAPH has been estimated to be 59 %. Risk factors associated with mortality include high level of mPAP, African American ethnicity and chronic respiratory failure requiring oxygen therapy [26].
Oxygen therapy should be prescribed if chronic hypoxaemia is present to prevent hypoxic vasoconstriction. The efficacy of immunosuppressive therapy on pulmonary vascular disease in sarcoidosis is not clear [13, 27, 28]. The use of PAH specific therapy in SAPH has predominantly been assessed in open-label observational studies [24, 29–32]. However, a recent multicenter, double-blind, randomized trial comparing bosentan versus placebo in 35 patients with sarcoidosis and concomitant precapillary PH showed improvements in hemodynamics (mPAP and pulmonary vascular resistance), but no effect on exercise capacity (6MWD) or functional class in patients treated with bosentan [33]. A recent meta-analysis of available studies of specific therapies in SAPH confirmed these data [34]. Gas exchange deterioration may also occur following vasodilator therapy via uncoupling of hypoxic pulmonary vasoconstriction, resulting in worsening of ventilation/perfusion mismatch [35]. Furthermore, potential risk of pulmonary edema can occur in cases with predominant venular involvement in a manner similar to PVOD [24, 36]. Thus, current guidelines do not support the use of PAH specific therapy in SAPH and off-label use of these therapies should only be considered in experienced PH centres.
Finally, because of the poor prognosis of SAPH and the lack of efficacy of specific PAH therapy, lung transplantation should be considered earlier in the course of the disease, despite the fact that the presence of PH prior to lung transplantation represents a risk factor of peri-transplant mortality [26] and primary graft dysfunction [37].
PH Associated with Pulmonary Langerhans’ Cell Histiocytosis
Langerhans’ cell histiocytosis is a systemic disease characterized by abnormal proliferation and infiltration of organs by Langerhans’ cells. Pulmonary Langerhans’ cell histiocytosis (PLCH) is a rare cause of diffuse parenchymal lung disease and occurs principally in smokers. PH can complicate the course of PLCH and severe PH is frequently reported in advanced disease [38–40]. Prevalence of severe PH (defined by mPAP ≥35 mmHg) in PLCH patients referred for lung transplantation assessment has been reported to range from 44 to 100 % [38, 39] significantly higher than other chronic lung diseases such as COPD or IPF [38]. Histopathological studies have shown a specific and diffuse pulmonary vasculopathy which predominantly involves the pulmonary veins and, to a lesser extent, the muscular pulmonary arteries (Fig. 33.2) [38]. Widespread pulmonary vascular lesions are present in the majority of PHLC related PH. Uncommonly, vascular lesions are due to direct infiltration by Langerhan’s cells [38]. Pulmonary vascular involvement is usually characterized by a proliferative vasculopathy with intimal fibrosis and medial hypertrophy involving the small to medium-sized pulmonary arteries and septal veins. Notably, significant venous involvement with a “veno-occlusive pattern” is present in up to one third of patients [38]. Vascular lesions may be observed in areas free from parenchymal lesions [38, 41].
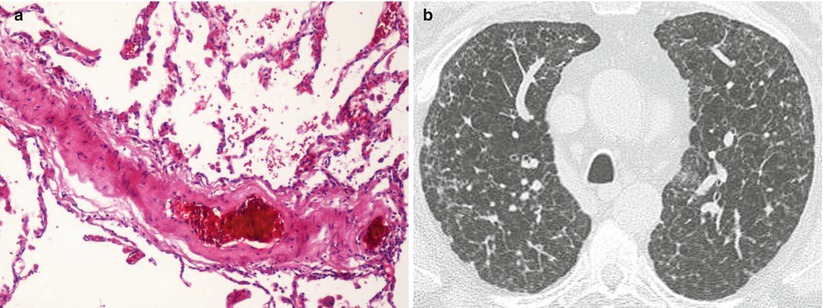
Fig. 33.2
Pathologic assessment and high-resolution CT of the chest of a patient with pulmonary hypertension associated with pulmonary Langherhans’ cell histiocytosis. (a) Diffuse pulmonary vasculopathy which predominantly involves the pulmonary veins and, to a lesser extent, the muscular pulmonary arteries. See intimal fibrosis of a septal vein with partial obliteration. Magnification 100, hematoxylin-eosin staining. (b) High-resolution CT of the chest showing multiple small cysts and nodules
Discrepancy between hemodynamic severity and lung parenchymal involvement is frequently observed in PLCH related PH. Indeed, hemodynamic parameters and pulmonary function tests are not correlated, suggesting that a specific pulmonary vascular involvement occurs independently of parenchymal lesions [38]. Hemodynamic impairment appears to be the main source of exercise limitation in PLCH related PH [42]. In a retrospective study of 29 patients with PLCH related PH, Le Pavec et al. [43] demonstrated severe haemodynamic impairment with 66 % of subjects displaying a mPAP ≥40 mmHg. In 23 of the 29 patients from this cohort, PH was diagnosed with a mean delay of 11 years after the initial diagnosis of PLCH. In this retrospective study, PAH specific therapies improved hemodynamics (mPAP and pulmonary vascular resistance) associated with an improvement in functional class in 2/3 of the patients and an increased of 6MWD >10 % in 45 % of patients. Functional class was the only predictor of death and the use of PAH specific therapy was not associated with improvement in survival [43]. PAH specific therapies appeared to be relatively safe in this context, nor significant worsening of gas exchange nor pulmonary edema related to potential venular involvement were reported in PLCH related PH. However, severe acute pulmonary edema has been reported by other authors during initiation of intravenous epoprostenol therapy [38, 41, 44]. Lungs or heart-lung transplantation remains the treatment of choice for end-stage PLCH and/or for severe PLCH related PH [39]. Of note, recurrence of PLCH in lung allografts has been reported and risk of recurrence was associated with the presence of extra-pulmonary disease prior to transplantation [39].
PH in Combined Pulmonary Fibrosis and Emphysema Syndrome
Combined pulmonary fibrosis and emphysema (CPFE) is a syndrome characterized by diffuse destruction of the lung parenchyma resulting from the combined effects of lung fibrosis predominant in the lower lobes and emphysema in the upper lobes [9, 45, 46]. Despite extensive parenchymal involvement, pulmonary function tests are often well preserved in this condition, but associated with marked reduction in the DLCO and severe hypoxemia. CPFE is classified in sub-group 3.3 “Other pulmonary diseases with mixed restrictive and obstructive pattern” (Table 33.1) [3]. CPFE is frequently associated with precapillary PH, usually with severe hemodynamic impairment [47–49]. The main assumption regarding the mechanism of PH in this context, is the association of alveolar destruction from emphysema and alveolar membrane thickening from fibrosis, leading to reduced lung perfusion and obliteration of the pulmonary vascular bed. The prevalence of PH in CPFE has been estimated to be 50 % in a retrospective study using echocardiography [46]. A retrospective study reported the haemodynamic, functional and survival characteristics of 40 patients dysplaying PH associated with CPFE [50]. At the time of diagnosis, right heart catheterization revealed moderate to severe PH (mPAP of 40 ± 9 mmHg, pulmonary vascular resistance of 521 ± 205 dyn·s−1·cm−5, and cardiac index of 2.5 ± 0.7 L.min−1.m2). Furthermore, 85 % of the patients were in either functional class III or IV, with a mean 6 min walking distance of only 244 ± 126 m. Prognosis was poor with 1-year survival estimated at 60 %. Univariate analysis found that DLCO <22 %, pulmonary vascular resistance >485 dyn/s/cm5, and cardiac index <2.4 L/min/m2 were predictive factors of death [50]. Interestingly, CPFE could also be present in connective-tissue diseases, with a similar prevalence of PH [51].
PAH-specific therapies are not recommended due to the lack of published evidence and the potential risk of aggravating hypoxemia by worsening ventilation/perfusion mismatch [35]. Finally, lung transplantation should be considered in selected patients and supportive care for chronic respiratory failure, especially oxygen therapy, is required.
PH Associated with Neurofibromatosis Type 1
Neurofibromatosis type 1 (NF1), also known as Recklinghausen’s disease, is a genetic disease, with an incidence of 1:2,500 births that has a typical clinical diagnosis: neurofibromas, cafe-au-lait spots, lentigines and squelletal lesions [52]. The respiratory involvement is rare and is almost exclusively represented by kysts, bulae or interstitial infiltrates that can go up to pulmonary fibrosis [52]. Initially pulmonary hypertension has been described as a consequence to an advanced parenchymal disease, but evidence show that in patients with minimal parenchymal involvement there is a plexiform pulmonary arteriopathy similar to that observed in idiopathic PAH [53, 54–57]. Recently we identified and published a series of 8 cases of NF1 associated PH, in patients with mild or absent lung parenchymal abnormalities [58]. These patients developed PH late in the course of NF1, with a median age at diagnosis of 62 years (range 53–68 years). Although in NF 1 the sex ratio is 1, there was a clear female predominance in NF 1 PAH patients as in idiopathic and heritable PAH. At time of diagnosis they had severe clinical, functional and hemodynamic impairment. Of the 8 reported cases, 3 had no significant lung involvement on HRCT. Spirometry was normal in 5 patients with DLCO markedly decreased in all patients as a sign of pulmonary vascular involvement. Lung transplantation was performed in one patient displaying severe parenchymal disease with diffuse interstitial fibrosis and widespread cystic changes. Pathologic assessment of explanted lungs confirmed interstitial fibrosis with partial loss of parenchymal architecture, but also showed pronounced pulmonary arterial remodeling (Fig. 33.3) [58]. For the patients in our cohort and the other case reports response to specific PH therapy was poor, which resulted in a global poor outcome [57–59].
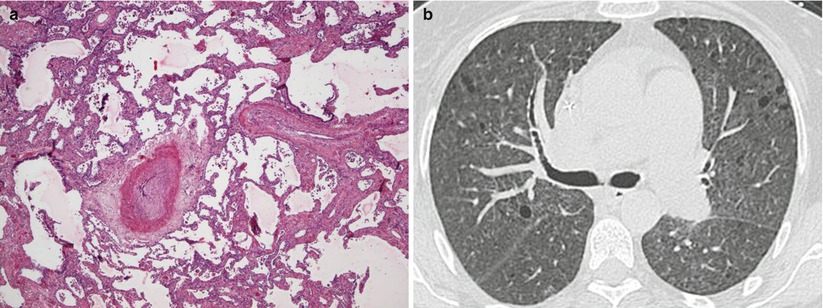
Fig. 33.3
Pathologic assessment and high-resolution CT of a patient with NF1-associated pulmonary hypertension. (a) Interstitial fibrosis with partial loss of parenchymal architecture associated with pronounced arterial remodeling complete occlusion by intimal fibrosis. Magnification 40, hematoxylin-eosin staining. (b) High-resolution CT of the chest showing diffuse small rounded lung cysts
In conclusion after all these new data, NF 1 related PH was put in group 5 in the updated clinical classification of pulmonary hypertensions, along other forms of PH with unclear/multifactorial mechanisms [60]. PH represents a rare but severe complication of NF1 that is characterized by a late onset, with female predominance, severe functional and hemodynamic impairment, and poor outcome. Specific PAH therapy seems to have only modest effect in these patients and these patients should be referred for lung transplantation if eligible.
PH Associated with Lymphangioleiomyomatosis
Lymphangioleiomyomatosis (LAM) is a rare multisystem disorder predominantly affecting women in their reproductive years [61]. It may occur sporadically with a prevalence of 1/400.000 adult females or in about 30 % of patients in the setting of tuberous sclerosis complex [61, 62]. LAM is characterized by an abnormal proliferation of smooth muscle like cells (also called LAM cells) along lymphatics in the lungs and abdomen that leads to diffuse cystic lung disease, recurrent pneumothoraces, benign renal tumours, pleural and peritoneal chylous effusions and abdominal lymphangioleiomyomas [62, 63]. At the pulmonary level, metalloprotease secretion by the LAM cells leads to the formation and progression of thin wall cysts, which in turn are responsible for airflow obstruction, low DLCO and chronic respiratory insufficiency [61–64]. The current accepted model for LAM is consistent with Knudson’s “Two-hit” hypothesis of tumour development: an initial mutation in either TSC1 or TSC2 is followed by a second hit represented by loss of heterozygosity, causing the loss of function of either TSC1 or TSC2 gene products [65]. A major role of the complex hamartin-tuberin is to inhibit mTOR, implicated in cell growth, indeed the mutation in both TSC1 and TSC2 induces activation of mTOR [62]. Recently, sirolimus, a mammalian target of rapamicyn (mTOR) inhibitor has been shown to slow the rate of lung function decline [66]. But even with all these new and promising data, lung transplantation remains the only option for patients with advanced respiratory disease [61, 67–69]. In the latest clinical classification of pulmonary hypertension, PH associated with LAM is included in group 5 with unclear multifactorial mechanisms [3]. There are two major hypotheses that can explain PH in LAM patients. The first is the classic hypoxic vasoconstriction mechanism which occurs in the context of severe parenchymal distortions caused by cysts [70]. The second is related to an up regulated mTOR secretion by the LAM cells, activation of mTOR complexes 1 and 2 in the context of hypoxia which in the end lead to vascular smooth cell proliferation and PH [71, 72].
Different studies evaluated the prevalence and characteristics of LAM related PH. By using cardiac echography with PH being defined as a systolic pulmonary arterial pressure >35 mmHg, Taveira-Dasilva et al. reported 8 cases of LAM related PH from 120 patients screened (prevalence 6.6 %) [70]. In a series of LAM patients referred for lung transplantation, by using right heart catheterization, Reynaud-Gaubert et al. found precapillary PH in 9/20 patients (prevalence 45 %) [67]. Based on these studies, the recent European Respiratory Society guidelines for the diagnosis and management of LAM has underlined that PH has not been reported frequently in LAM patients and that screening for PH is not recommended in patients with non-severe LAM [61]. However, it was suggested that estimation of PAP should be performed in patients considered for lung transplantation [68].
In the most recent retrospective multicenter study, Cottin et al. reported 20 patients with LAM and precapillary PH confirmed by right heart catheterization [73]. The mean age at diagnosis of PH was 49 years with a mean time interval between LAM and PH diagnosis of 9.2 years [73]. Hemodynamics showed moderate PH with a mPAP of 32 ± 6 mmHg, a cardiac index of 3.5 ± 1.1 l · min−1 · m−2 and pulmonary vascular resistance of 376 ± 184 dyn · s · cm−5, with only 4 patients (20 %) having a mean PAP > 35 mmHg [73]. These hemodynamic results suggested that in the majority of cases, PH was mild or moderate and related to the severity of pulmonary involvement [73]. Pulmonary function tests showed a decreased FEV1 at 42 ± 25 % and DLCO at 29 ± 13 % with blood gases showing mild hypoxemia (PaO2 of 7.4 ± 1.3 kPa in room air) [73]. Only 6 patients received oral PAH specific therapy and showed a decrease in m PAP and pulmonary vascular resistance from baseline. The authors showed that the overall probability of survival at 2 years was 94 % [73].
In conclusion, mild to moderate PH is relatively a common finding in LAM patients with severe lung involvement with chronic hypoxemia and pulmonary capillary destruction caused by cystic lung lesions probably may represent the predominant mechanism of PH in this setting. Nevertheless, some patients may have specific pulmonary vascular involvement, and one potential mechanism may be the activation mTOR, as proposed in patients with NF1-associated PH. These patients may be candidates for specific PAH therapies, but further studies are needed in order to assess this possibility.
Hereditary Hemorrhagic Telangiectasia
Hereditary hemorrhagic telangiectasia (HHT) is characterized by mucocutaneous telangiectases, recurrent epistaxes, macroscopic arteriovenous malformations (particularly in the pulmonary, hepatic, and cerebral circulation) and more rarely PAH. HHT is inherited in an autosomal dominant fashion with late-onset penetrance and nearly complete penetrance (97 %) at the age of 60 years. Several genes have been implicated in the pathogenesis of HHT, including activin receptor-like kinase-1 (ACVRL1 or ALK-1) located on chromosome 12, endoglin on chromosome 9. ACVRL1 and endoglin genes are involved in the transforming growth factor-ß (TGF-ß) signalling pathway as BMPR2 gene, the main predisposing gene of heritable PAH. Indeed, proteins encoded by BMPR2 and ACVRL1 genes, form an heteromerix complex, associated with endoglin [74].
Several mechanisms may lead to PH in HHT patients. Arteriovenous malformations may create clinically significant right-to-left shunts, causing hypoxemia, paradoxical embolism, stroke, and cerebral abscesses [75–79]. PH may develop as a consequence of a hyperkinetic state resulting in heart failure associated with high cardiac output. However, HHT is also associated with PAH characterized by remodeling of small pulmonary arteries, with broadly similar histologic lesions than observed in idiopathic PAH [79–81]. Many case series have reported the association of ACVRL1 mutations and PAH in HHT patients without any other cause of PH [80–85]. At the opposite, only few cases of PAH in endoglin mutants have been reported, although endoglin and ACVRL1 mutations are present in a comparable proportion in the population, suggesting a less potent association between endoglin and PAH [82, 83, 86]. We previously demonstrated that PAH patients carriers of an ACVRL1 mutation are significantly younger (21.8 ± 16.7 years) at PAH diagnosis, as compared to BMPR2 mutation carriers (35.7 ± 14.9 years) and non-carriers (47.6 ± 16.3 years) with a more rapid disease evolution [80]. Interestingly, ACVRL1 mutation carriers may develop severe PAH without any clinical evidence of HHT because of the early development of PAH in these patients and the late-onset penetrance of ACVRL1 mutations for HHT manifestations [80]. In conclusion, the absence of HHT clinical manifestation in PAH patients should not exclude the diagnosis of PAH associated with ACVRL1 mutation and a detailed familial history and a careful examination of PAH patients and first-degree relatives for stigmata of HHT may help detect these patients.
Pulmonary Veno-Occlusive Disease
Pulmonary veno-occlusive disease (PVOD) and pulmonary capillary hemangiomatosis (PCH) are uncommon forms of PH difficult to diagnose [87]. It has been suggested that PVOD represents 5–10 % of histological forms of cases initially considered as idiopathic PAH [5, 6, 88, 89]. Whereas idiopathic PAH shows a distinct female preponderance, PVOD is characterised by a higher male/female ratio [6]. Initially, PVOD and PCH were placed in two different groups, both distinct from PAH. Indeed, histological examination of lung samples shows extensive and diffuse occlusion of pulmonary veins by fibrous tissue and intimal thickening involving preferentially venules and small veins in lobular septa in PVOD (Fig. 33.4), and localized capillary proliferations which could obstruct veins and venular walls in PCH [8, 90]. Similarities in pathological features and clinical presentation suggested that PVOD and PCH may be two presentations of the same disease. According of these observations PVOD/PCH was grouped in a subgroup of group 1 in the recent classification of PH (group 1′) (Table 33.1). Indeed, a clinicopathologic study analyzing specimens from 35 patients diagnosed as having either PVOD or PCH [8]. The authors concluded that lesions of capillaries were present in 3/4 of cases diagnosed as PVOD and that significant venous involvement was present in 4/5 cases initially diagnosed as PCH. It has been hypothesized that capillary hemangiomatosis may result from an angioproliferative process associated with venous obstruction, as observed in PVOD.