Introduction
Pulmonary hypertension is defined as a mean pulmonary artery pressure of greater than 25 mm Hg at rest or 30 mm Hg with exercise. The term pulmonary arterial hypertension (PAH) denotes a series of apparently unrelated disorders many of which share the histopathologic entity of plexogenic pulmonary arteriopathy (PPA). Typically in PAH the mean pulmonary capillary wedge pressure is less than 15 mm Hg.
Examples of PAH include idiopathic PAH, familial PAH and pulmonary hypertension associated with scleroderma, hepatic cirrhosis, HIV infection and Eisenmenger’s syndrome. It has also been described in association with ingestion of slimming agents, e.g dexfenfluramine, and with other unusual conditions including pulmonary venoocclusive disease and glycogen storage disorders (Box 70.1).
Pulmonary hypertension can also occur in association with cardiac and respiratory diseases. Examples of the former include left ventricular failure, chronic left atrial hypertension and mitral valve disease and of the latter chronic obstructive pulmonary disease (COPD), interstitial lung disease, disorders of pulmonary development and sleep apnea syndrome. Chronic thromboembolic pulmonary hypertension (CTEPH) affecting proximal or distal pulmonary arteries may occur for unknown cause or may be associated with thrombophilia abnormalities such as protein S and C deficiency or abnormalities in factor V. Prothrombotic states can also accompany connective tissue disorders or malignancy. In addition, pulmonary hypertension can result from other unusual conditions, e.g. lymphan-gioleiomyomatosis (see Box 70.1).
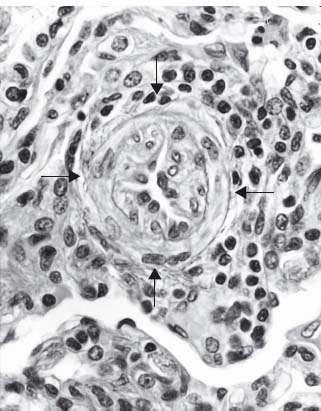
Idiopathic PAH was initially described by Romberg as “sclerosis of the pulmonary arteries” over 100 years ago.1 Dresdale and colleagues termed the condition primary pulmonary hypertension some 50 years later.2 Wood observed a reduction in pulmonary arterial pressure in response to intravenous administration of acetylcholine and suggested that a “vasoconstrictive factor” was responsible for its pathogenesis.3 Subsequently clinical experience with vasodilator therapy was disappointing and this was consistent with reports from Wagenvoort and Wagenvoort4 (4) and Caslin and colleagues which suggested that more extensive vascular injury and remodeling occurred in this process (Fig. 70.1).5–7 Over the years that followed the term PAH was defined as an umbrella term to link a number of apparently different disease processes which shared similar his-topathologic appearances and were associated with severe elevation in pulmonary vascular resistance (PVR). Authors described pulmonary hypertension occurring in association with ingestion of aminorex fumarate (an appetite suppressant) in Western Europe,8 in association with connective tissue disease,9 hepatic cirrhosis and portal hypertension10 and with HIV infection.11 Traditionally idiopathic PAH was described in young females although with increasing awareness, the condition is now diagnosed in patients beyond the fourth and fifth decades of life. The incidence and prevalence of idiopathic PAH are estimated to be four per million and ten per million of the population respectively. Overall, the prevalence of PAH is estimated to be in the region of 100 per million of the population. The incidence and prevalence of pulmonary hypertension in patients with cardiac and respiratory disorders are not precisely known although they are believed to be considerably higher than for PAH. PAH is associated with a poor survival and a poor quality of life. There is no cure, limited treatment options and incomplete understanding of the disease.12,13
Figure 70.1 Pulmonary arteriole from a patient with PAH showing extensive reduction in vessel lumen as a consequence of smooth muscle cell migration from the inner half of the media. Once in the lumen, the cells become myofibroblasts and proliferate. Note also a surrounding inflammatory infiltrate (H&E, medium power).
BOX 70.1 Diagnostic classification of pulmonary hypertension (World Conference on Pulmonary Hypertension, Venice 2003)
Pulmonary arterial hypertension (PAH)
- Idiopathic PAH
- Familial PAH
- Related to:
- Connective tissue diseases
- HIV
- Portal hypertension
- Anorexigens
- Congenital heart diseases
- Pulmonary capillary hemangiosis
- Pulmonary veno-occlusive disease
- Others (e.g. glycogen storage disease, splenectomy)
Associated with left heart disease
- Atrial or ventricular dysfunction
- Valvular disease
Associated with lung disease/hypoxemia
- COPD
- Interstitial lung diseases
- Sleep-disordered breathing
- Developmental abnormalities
- Chronic exposure to high altitude
Associated with chronic thrombotic and/or embolic disease
- Obstruction of proximal pulmonary artery
- Obstruction of distal pulmonary artery
- Non-thrombotic pulmonary emboli (e.g. tumor)
Miscellaneous
- Histiocytosis
- Lymphangioleiomyomatosis
- Sarcoidosis
- Compression of pulmonary vessels (adenopathy, tumor, mediastinal fibrosis)
PPA occurs in a select group of disorders. It is unclear why this histopathologic entity occurs although it is possible that the lung has only a finite number of responses to injury which feed into final common pathway mechanisms. This may explain why similarities occur in patients with conditions such as obliterative bronchiolitis following lung transplantation and those with obliterative bronchiolitis associated with rheumatoid lung disease or respiratory syncytial virus infection in childhood. Similarly many conditions have been implicated as causative in the acute respiratory distress syndrome yet the pathology is similar regardless of etiology.
In PPA the pathophysiologic triggers are not clear. Following initial vasoconstriction smooth muscle migration occurs from the inner half of the media of muscular pulmonary arterioles into the vessel lumen where these cells become myofibroblasts capable of laying down either smooth muscle or fibrous tissue. Once in the lumen, the cells proliferate in a concentric fashion and ultimately obliterate the lumen (see Fig. 70.1). As this process develops the radius of the vessel lessens and in accordance with Poiseuille’s law, the resistance to flow increases. When sectioned, these vessels have the appearance under the microscope of a cut onion and the process has been described as “onion skin proliferation”. Interestingly, in COPD smooth muscle cells migrate into the vessel lumen but migrate in a longitudinal fashion and although reduced, there is less compromise to the radius than with PPA. At proximal points of weakness in the vessel (often at areas of branching), the vessel distends and ruptures. Hemorrhage follows and primitive blood vessels grow into this area in a haphazard or plexiform arrangement. The combination of concentric laminar intimal (“onion skin”) proliferation and plexiform lesions is referred to as PPA. Some authors believe that plexiform lesions represent a type of collateral circulation.
It is not clear why these particular changes occur in diseases with such diverse etiology and clinical presentation. Immunoreactive cells in the lung for gastrin-releasing peptide and calcitonin may be important factors in the smooth muscle cell migration process.14 Endothelial injury as a consequence of damage by toxins or genetic factors may also be important and indeed, overexpression of endothelin-1 and reduced levels of prostacyclin synthase have been noted in plexiform lesions.15–20 There is ongoing extensive research into endothelial dysfunction in patients with PAH.
The natural history of idiopathic PAH has been described and the National Institute of Health Registry followed up 194 patients with this condition enrolled at 32 medical centers between 1981 and 1985. The median survival was 2.8 years with one-, three- and five-year survival rates of 68%, 48% and 34% respectively21 Indeed, actuarial five-year survival for untreated patients with PAH who are in class IV New York Heart Association (NYHA) status is significant lower than that for patients with lung, breast, prostate, colon and gastric carcinoma. The following factors are useful in predicting mortality in PAH:
- etiology
- unctional capacity (NYHA or PAH class)
- xercise capacity (unencouraged six-minute walk test)
- hemodynamics (severity of right ventricular dysfunction)
- echocardiographic parameters.
It appears that survival for patients with PAH associated with scleroderma is worse than for patients with idiopathic PAH22 while survival for patients with PAH associated with HIV infections is similar to those patients with idiopathic PAH.23 Interestingly, in a number of studies most deaths in patients with HIV infection and PAH were related to PAH. Patients with congenital heart disease may have a better prognosis than those with idiopathic PAH although further experience is necessary to validate this.24
As in adults, the prognosis in children with PAH is linked to the underlying etiology.
The incidence and prevalence of pulmonary hypertension in patients with cardiac and respiratory disorders are not precisely known but are believed to be considerably higher than for the causes of PAH. In these conditions expert medical treatment focusing on the underlying cardiac, pulmonary or hematologic abnormalities is key to the management of the co-existing pulmonary hypertension, at least in the first instance.
Higher NYHA functional class (III or IV) is associated with increased mortality in both treated and untreated patients with idiopathic PAH.21,22,25 Failure to improve NYHA functional class or deterioration in NYHA functional class while on therapy may of itself be predictive of a poor outcome. The unencouraged six-minute walk test is an easy-to-perform and reproducible modality of assessing exercise capacity in patients with PAH and may be an independent predictor of survival for these patients. One study suggested that a six-minute walk test of less than 332 meters was associated with a significantly lower survival rate than for patients whose six-minute walk test exceeded this distance.26
In patients with suspected pulmonary hypertension right heart catheterization is performed to confirm the presence and severity of pulmonary hypertension and to help establish the underlying diagnosis. Furthermore, right heart catheterization is required to guide therapeutic intervention. The PVR (mm Hg/L/min) is calculated as follows:

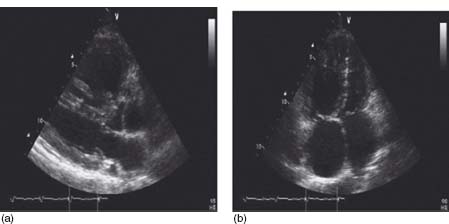
Echocardiography is an integral part of the evaluation of a patient with pulmonary hypertension. Common findings in patients with PAH include right atrial and right ventricular enlargement, reduced right ventricular function, paradoxic movement of the intraventricular septum and tricuspid regurgitation (Fig. 70.2). The presence of a pericardial effusion seems to be associated with a poorer prognosis.27 Another indicator of an adverse outcome is an elevated Doppler echocardiography right ventricular index.28
Figure 70.2 Two-dimensional echocardiography from a patient with PAH showing right ventricular enlargement and paradoxic encroachment of the intraventricular septum from the right ventricle into the left ventricle. (a) parasternal long axis view. (b) Apical four-chamber view.
Correlation between estimated pulmonary artery systolic pressure on echocardiography and at right heart catheterization is not always close. Echocardiographic measurements are usually dependent on assessment of the tricuspid regurgitant velocity and errors with this measurement and with subsequent formulae employed often contribute to inaccuracy.
Patients with PAH have similar quality of life scores when compared with those for patients with COPD and with end-stage renal failure.
Early on in the course of their illness, patients with PAH may be asymptomatic or experience dyspnea with exertion. In the early stages of the disease the non-specific nature of the symptoms may lead to either failure of diagnosis or incorrect diagnosis. Many patients have had their symptoms attributed to depression. As the condition progresses, the PVR rises and the cardiac output falls. At this stage patients may change from having relatively few symptoms to experiencing dyspnea, palpitations, chest pain (right ventricular angina), presyncope or syncope. Initially these symptoms may occur with exertion and subsequently at rest. As the condition progresses further peripheral edema, ascites, plethora and more profound fatigue develop as right ventricular dysfunction and tricuspid regurgitation evolve. Ultimately right heart failure and death occur.12
Disease progression and response to therapy can be functionally assessed using the World Health Organization classification of functional capacity which is an adaptation of the NYHA system (Table 70.1).
Table 70.1 WHO classification of functional status of patients with pulmonary hypertension (PH)*
| Class | Description |
I | Patients with PH in whom there is no limitation of usual physical activity; ordinary physical activity does not cause increased dyspnea, fatigue, chest pain or presyncope |
II | Patients with PH who have mild limitation of physical activity. There is no discomfort at rest, but normal physical activity causes increased dyspnea, fatigue, chest pain or presyncope |
III | Patients with PH who have a marked limitation of physical activity. There is no discomfort at rest, but less than ordinary activity causes increased dyspnea, fatigue, chest pain or presyncope |
IV | Patients with PH who are unable to perform any physical activity and who have signs of right ventricular failure. Dyspnea and/or fatigue may be present at rest, and symptoms are increased by almost any physical activity |
* Rich S. Primary Pulmonary Hypertension: Executive Summary. Evian, France: World Health Organisation, 1998.
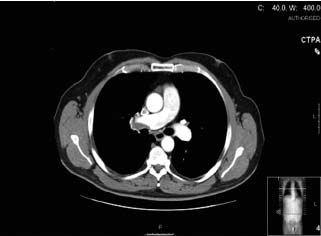
Since PAH can occur in association with other conditions, symptomatic evidence of a related illness should be considered. For example, orthopnea and paroxysmal nocturnal dyspnea suggest elevated pulmonary venous pressure and pulmonary congestion due to left-sided heart disease or pulmonary veno-occlusive disease. Features such as Raynaud’s phenomenon or arthralgias or rash may suggest the possibility of an underlying connective tissue disease. Other lung diseases such as lung fibrosis, COPD and obstructive sleep apnea can be associated with pulmonary hypertension and will need to be assessed and potentially treated in addition to the primary lung disease. One should also exclude pulmonary thromboembolic disease by performing either a ventilation/perfusion (V/Q) or computed tomographic pulmonary angiography (CTPA) scan (Fig. 70.3) or pulmonary angiography. For many patients with pulmonary hypertension associated with other conditions, treatment of the primary condition will significantly improve their pulmonary hypertension.
Figure 70.3 CTPA from a patient with pulmonary hypertension associated with pulmonary thromboembolic disease.
The clinical features of pulmonary hypertension can be subtle and may be missed on physical examination. Important features to identify are a loud pulmonary component to the secondary heart sound which reflects increased force of pulmonary valve closure secondary to elevated pulmonary artery pressure. Other auscultatory abnormalities include an early systolic ejection click due to sudden interruption of pulmonary valve opening, a midsystolic ejection murmur caused by turbulent transvalvular pulmonary flow and a right ventricular fourth heart sound. Additional features include a palpable left parasternal lift as a consequence of right ventricular hypertrophy and a prominent jugular “a” wave which suggests high right ventricular filling pressures. As the condition develops further a diastolic murmur of pulmonary regurgitation may be noted together with a pansystolic murmur of tricuspid regurgitation. Tricuspid regurgitation may also be detected by inspection of the neck demonstrating an elevated jugular venous pressure with accentuated V waves, a hepatojugular reflux and a pulsatile liver. Right ventricular failure is associated with a right ventricular third heart sound, distension of the jugular veins, pulsatile hepatomegaly, ascites and peripheral edema. As the disease progresses hypotension and poor peripheral perfusion indicate significant reduction of cardiac output with elevated peripheral vascular resistance. Cyanosis is noted in 20% of patients with idiopathic PAH; clubbing is rare.12 Other cardiac and respiratory abnormalities may give further clues to other diagnoses and in particular, one must exclude CTEPH and potential underlying causes of this condition.
In addition to routine hematologic and biochemical indices (full blood count, urea, creatinine and electrolytes, liver function test), one should screen for autoimmune diseases and clotting abnormalities and check arterial blood gas analysis on room air. Genetic screening is also important. A chest X-ray may show enlarged main and hilar pulmonary arteries, peripheral pruning of pulmonary vessels and cardiomegaly of right ventricular configuration. ECG may be normal, may identify right axis deviation and right ventricular strain or show evidence of right atrial or right ventricular hypertrophy or arrhythmias. Two-dimensional echocardiography can provide useful information of right and left heart function and valvular abnormalities and can provide an estimate of the pulmonary artery systolic pressure. Ventilation perfusion and CTPA scanning can help exclude CTEPH and right heart catheterization can help confirm the presence of pulmonary hypertension and provide important information as to its origin. Full lung function tests may be normal or show abnormalities consistent with other diagnoses, e.g. low transfer factor for patients with lung fibrosis, left heart failure or CTEPH.
Evidence for diagnostic tests in patients with suspected pulmonary arterial hypertension
As PAH is a relatively rare condition, the literature is lacking in large series of appropriately conducted clinical trials which help establish firm guidelines for diagnosis and treatment. Furthermore, there is no effective cure and many treatments available will at best reduce the rate of deterioration of the illness and improve quality of life. Additionally, there is an important cost implication, particularly for long-term use of prostanoids and endothelin receptor antagonists. Therapies such as lung transplantation are limited by donor organ availability. Finally PAH represents a series of diverse conditions which interface at the level of the pulmonary circulation. Efforts to rationalize diagnostic modalities and treatment options are clearly difficult.
As a consequence, a number of expert consensus group meetings were organized to obtain broad agreement as to how patients with PAH should be diagnosed and treated.13 What is reported in this chapter represents a distillation of current international opinion. Often such expert opinion is based on clinical experience in conjunction with results obtained from clinical trials. Many of these trials provide encouraging results but may be criticized because of design, duration and relatively small numbers of patients. In the light of early results further prospective trials are under way. It should also be stressed that there are currently no firm data based on properly constructed clinical trials to advocate specific management strategies for patients whose pulmonary hypertension is associated with primary cardiac and pulmonary diseases.
Consensus of opinion recommends that for patients with suspected PAH routine hematologic and biochemical investigations be performed and that autoimmune profile and HIV status is assessed (Class I). Chest X-ray is considered routine as is 12-lead ECG. Doppler echocardiography is a good screening tool to detect PAH (Class I) and is useful at assessing morphology and abnormalities and in estimating right ventricular and pulmonary arterial systolic pressures (Class IIa), to assess left ventricular function (Class I) and (with contrast) intracardiac shunting (Class IIa).
V/Q scan or CT pulmonary angiography is recommended to exclude chronic pulmonary thromboembolic disease (Class IIa) and pulmonary angiography is advocated prior to considering pulmonary thromboendarterectomy (Class I). Lung function tests and arterial blood gas analysis are routinely performed (Class IIa). Estimate of gas transfer is useful in patients with PAH and scleroderma (Class IIa) and chronic pulmonary thromboembolism (Class IIa). Lung biopsy is not routinely advocated (Class I). Right heart catheter (Box 70.2) is required to confirm the presence of pulmonary hypertension, establish the specific diagnosis and determine its severity (Class I) and also to help guide treatment (Class IIa). Genetic testing and counseling should be offered to patients with familial PAH (Class I
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


