Pulmonary Hypertension: Introduction
Pulmonary hypertension (PH) is a hemodynamic abnormality common to a variety of conditions that is characterized by increased right ventricular (RV) afterload and work. The clinical manifestations, natural history, and reversibility of pulmonary hypertension depend heavily on the nature of the pulmonary vascular lesions and the etiology and severity of the hemodynamic disorder, although individual variability exists. For example, subacute or chronic hypoxia predominantly causes increased muscularization of the small muscular pulmonary arteries and arterioles with the intima relatively intact. Relief of the hypoxia improves or occasionally reverses the process with little or no pathologic residue.1,2 In contrast, the lesions of systemic sclerosis (scleroderma), mostly confined to the intima of the small pulmonary arteries and arterioles, are usually progressive and irreversible. Unlike these two examples, which spare the pulmonary capillary bed, the pulmonary capillaries are the primary site of involvement in pulmonary capillary hemangiomatosis.3
Because of its large capacity, its great distensibility, its low resistance to blood flow, and the modest amounts of smooth muscle in the small arteries and arterioles, the pulmonary circulation is not predisposed to become hypertensive. In normal individuals lying supine, systolic pressure is approximately 15 to 25 mm Hg; the corresponding diastolic pressure is 5 to 10 mm Hg. The mean driving pressure (ie, the difference between the mean blood pressure in the pulmonary artery and in the left atrium) is approximately 10 to 12 mm Hg. Because blood flow (cardiac output) is the same in both circulations in the absence of any systemic to pulmonary communications, the pulmonary vascular resistance (PVR) is approximately one-eighth of systemic vascular resistance. The large cross-sectional surface area of the pulmonary circulation, coupled with the distensibility of its thin-walled vessels and the large recruitable vascular reserve, account for these unique characteristics. During exercise, as pulmonary blood flow increases, new regions of the pulmonary vascular bed are open and existing vasculature dilates; accordingly, the pulmonary circulation is capable of accommodating a four-fold or greater increase in resting blood flow with virtually no change in pulmonary artery pressure, with a concomitant decrease in PVR.
When total cross-sectional area is decreased by destruction or obliteration of lung tissue or occlusive lesions in the resistance vessels, pulmonary arterial pressures increase. The degree of PH that develops is a function of the amount of the pulmonary vascular tree that has been eliminated. PH is most often associated with cardiac or pulmonary diseases. Although idiopathic pulmonary arterial hypertension (IPAH; formerly known as primary pulmonary hypertension [PPH]) is uncommon, it is well recognized as a distinctive clinical entity in which intrinsic pulmonary vascular disease is free of the complicating features of PH contributed by diseases of the heart and/or lungs. Mild PH can exist for a lifetime without becoming evident clinically. For example, native residents at high altitude, in whom mild to moderate PH is a natural result of sustained exposure to hypoxia, can adapt and function normally. When PH does become manifest clinically, the symptoms tend to be nonspecific (Table 71–1).
Definitions
PH is defined as a mean pulmonary arterial hypertension at least 25 mm Hg at rest measured by invasive monitoring. Pulmonary arterial hypertension (PAH) is defined as PH with normal left-sided filling pressure (ie, pulmonary capillary wedge pressure [PCWP] <15 mm Hg) and an increased PVR. PAH can be either acute or chronic. The acute form is usually a result of either pulmonary embolism (see Chap. 72) or the adult respiratory distress syndrome. This chapter deals with chronic PAH.
Pulmonary venous hypertension is usually encountered as a consequence of left ventricular (LV) dysfunction or mitral valve disease. Occasionally, it may occur in the course of fibrosing mediastinitis. The hallmarks of pulmonary venous hypertension are pulmonary congestion and edema. For practical purposes, pulmonary venous hypertension exists when pulmonary venous (or left atrium [LA]) pressure rises above 15 mm Hg. To maintain forward blood flow, pulmonary arterial pressure increases; however, this does not necessarily result in an increase in PVR. Additionally, PH can be mixed pulmonary vasculopathy, defined as PH with a PCWP >15 mm Hg and an increased PVR.
Cor pulmonale signifies the presence of PH and cardiac dysfunction in the setting of diseases affecting the structure or function of the lung.4 PH in patients with chronic lung disease tends to be less severe than in connective tissue diseases, chronic thromboembolic disease, or IPAH. PH may be severe, however, in some patients with interstitial lung disease.
Normal Pulmonary Circulation
Immediately before birth, pulmonary and systemic arterial blood pressures are near equal and approximately 70/40 mm Hg, with a mean of 50 mm Hg. Immediately after birth, with closure of the ductus arteriosus and initiation of ventilation, pulmonary arterial pressure falls rapidly to approximately one-half of systemic levels. Thereafter, pulmonary arterial pressures gradually decrease over several weeks to reach adult levels.5
In some neonates, the normal PH of the fetus fails to recede normally, generally as a result of either a developmental anomaly or a relentless increase in pulmonary vascular tone. In such infants, the persistent PH and RV failure may become life threatening. Surgical intervention or temporizing measures, such as the use of inhaled nitric oxide (NO) or extracorporeal membrane oxygenation, have improved the outcome for many infants with reversal of the pulmonary vascular abnormalities.6
In the normal adult at sea level, the small muscular arteries and arterioles in the lungs are thin-walled and contain very little smooth muscle. In contrast, in the fetus or the adult who has lived under hypoxic conditions (eg, native residents at high altitude), the media of the arterioles are thickened, and the muscle extends distally into precapillary vessels that are ordinarily devoid of muscle; that is, the precapillary vessels undergo remodeling.7
In addition to its role as a semipermeable barrier between blood and interstitium, the endothelium provides many biologically important functions, the net effect of which is the processing of blood flowing through the lungs. Among these functions are the synthesis, uptake, storage, release, and metabolism of vasoactive substances; transduction of bloodborne signals; modulation of coagulation and thrombolysis; regulation of cell proliferation; engagement in the local inflammatory and proliferative reactions to injury; involvement in immune reactions; and angiogenesis (see Chap. 5). Some of the enzymes involved in these processes, such as the angiotensin-converting enzyme, are found on the surface of endothelial cells; others, such as 5-nucleotidase, are found within the cell. Hence it is appropriate to regard the endothelium as an organ with diverse metabolic and endocrine functions, one that is unique because of its strategic location as a continuous, monolayered lining of blood vessels throughout the body. Importantly, the lungs contain the largest expanse of endothelium in the body.
The cells that make up the monolayered endothelial lining communicate not only with each other by anatomic junctions and bridges, but also with the underlying smooth muscle by way of biologically active substances. This interaction participates in regulating normal vasomotor tone as well as responding to the administration of vasoactive substances. Thus damage to the lining cells, proliferation of the intima, or hypertrophy of the smooth muscle will each upset the normal interplay.
For the adult pulmonary circulation, the definition of normal depends on the altitude. Table 71–2 compares the normal pulmonary hemodynamics of adults residing at sea level and above sea level. At sea level, a cardiac output of 5 to 6 L/min is associated with a pulmonary arterial pressure of approximately 20/12 mm Hg, with a mean of approximately 15 mm Hg. At an altitude of 15,000 ft, the same level of blood flow is associated with somewhat higher pressures (see Table 71–2). Pulmonary arterial pressures also tend to increase somewhat with age.
| Sea Level | Altitude (~15,000 ft) | |
|---|---|---|
| Pulmonary arterial pressure (PPA), mm Hg | 20/12, 15 | 38/14, 25 |
| Cardiac output (Q), L/min | 6 | 6 |
| Left atrial pressure (PLA), mm Hg | 5 | 5 |
| Pulmonary vascular resistance (PVR),a (mm Hg/L)/min (R units) | 1.7 | 3.3 |
A pressure drop of only 5 to 10 mm Hg between the pulmonary artery and left atrium accompanies the cardiac output of 5 to 6 L/min (see Table 71–2). Determination of PVR, calculated as the ratio of the difference in mean pressure at the two ends of the pulmonary vascular bed (pulmonary arterial pressure minus LA pressure) divided by the cardiac output (see Table 71–2), is a practical clinical tool for assessing the hemodynamic state of the pulmonary circulation and for distinguishing between active and passive changes in the pulmonary resistance vessels (eg, the effect of administering a vasodilator agent to a patient with PH). In practice, because the LA may not be readily accessible, pulmonary capillary wedge pressure is generally substituted for LA pressure.
Another approach to defining certain characteristics of the pulmonary arterial tree and its behavior—that is, elastic properties and geometry—is the calculation of pulmonary arterial input impedance. This approach has more physiologic than clinical value. It takes into account the pulsatile nature of pulmonary arterial pressures and flow. Like vascular resistance, it is defined as a ratio. But instead of a ratio involving mean pressures and blood flow, the ratio uses the amplitudes of pulsatile pressure to oscillatory flow near the beginning of the pulmonary artery at a particular frequency. Values for the ratio are calculated by compartmentalizing the pulsatile pressure and flow curves into their sinusoidal components.
PVR has proved useful in assessing the state of the normal and abnormal pulmonary circulation, but reliance on the calculated resistance must be done with caution. For example, PVR may decrease despite an increase in pulmonary arterial pressure if cardiac output increases to a greater degree than the increase in pulmonary arterial pressure as a result of a drug-induced primary effect on cardiac contractility or heart rate with little direct effect on pulmonary vasomotor tone. It is unlikely that this pattern of hemodynamic response will be beneficial. Also, a clinical shortcut, such as the substitution in the numerator of the pulmonary arterial pressure for the pressure drop between the pulmonary artery and LA, may be useful empirically but deprives the calculation of any physiologic meaning. Finally, the clinical significance of a value calculated for PVR depends heavily on the implications of the hemodynamic changes on the work of the RV. For example, the same decrease in calculated PVR brought about by two different pulmonary vasodilators may affect the work of the RV differently: Should one agent elicit a decrease in pulmonary arterial pressure along with an increase in cardiac output (an ideal response), it is more apt to be of long-term benefit than another agent that, although increasing the cardiac output, fails to decrease the pulmonary arterial pressure.
In the normal lung, a considerable increase in cardiac output, that is, three to five times that at rest, generally increases pulmonary arterial pressure by only a few millimeters of mercury with a concomitant decrease in PVR. On the other hand, in pulmonary hypertensive states, in which the distensibility and recruitability of the pulmonary vascular bed are restricted by disease, pulmonary arterial pressure increases along with even small incremental increases in pulmonary blood flow. Changes in pulmonary blood volume are much more subtle than changes in blood pressure or flow in their hemodynamic effects; they are also much more difficult to quantify. Clinical clues can be helpful in recognizing that the pulmonary blood volume has increased. Often a fullness of the pulmonary vascular pattern on the chest radiograph along with evidences of interstitial edema suggest that pulmonary blood volume has increased acutely. In chronic mitral stenosis or LV failure, the pulmonary blood volume is not only increased, but is also redistributed toward the apices of the lungs, that is, cephalization (see Chap. 17).
Autonomic innervation of the pulmonary vascular tree plays a lesser role in modulating vasomotor tone than do local stimuli, particularly hypoxia. Indeed, hypoxia can exert its pulmonary pressor effect in the denervated lung. The mechanism by which hypoxia exerts its local pressor effect is not fully characterized, but appears to involve altered smooth muscle cell membrane ion channel activity.2 Acidosis potentiates the hypoxic pressor effect. Hypercapnia also exerts a pulmonary pressor effect due to the respiratory acidosis that it generates.
Pulmonary Hypertension
PH is a final common hemodynamic consequence of multiple etiologies and diverse mechanisms. Among the underlying causes of PH are mechanical compression and distortion of the resistance vessels of the lungs (eg, by diffuse pulmonary fibrosis), hypoxic vasoconstriction (eg, in severe obstructive airways or diffuse parenchymal diseases), intravascular obstruction (eg, thromboemboli or tumor emboli), and combinations of mechanical and vasoconstrictive influences. The significance of PH, however, is that the increased afterload compromises RV function. Once pulmonary arterial pressures reach systemic levels, RV failure becomes inevitable.
The classification of PH has gone through a series of changes since the first classification was proposed in 1973 at an international conference on PPH endorsed by the World Health Organization. The most recent classification, the Dana Point 2008 4th World Symposium on Pulmonary Hypertension,8 is based on shared pathologic, pathobiologic, and clinical features (Table 71–3). PH patients can belong to more than one group; additionally, patients in Group 1 PAH can have more than one risk factor or associated condition. The terms PPH and secondary PH have been replaced with more specific and descriptive terms.
| 1. Pulmonary arterial hypertension |
| 1.1. Idiopathic PAH |
| 1.2. Heritable |
| 1.2.1. BMPR2 |
| 1.2.2. ALK1, endoglin (with or without hereditary hemorrhagic telangiectasia) |
| 1.2.3. Unknown |
| 1.3. Drug- and toxin-induced |
| 1.4. Associated with: |
| 1.4.1. Connective tissue diseases |
| 1.4.2. HIV infection |
| 1.4.3. Portal hypertension |
| 1.4.4. Congenital heart diseases |
| 1.4.5. Schistosomiasis |
| 1.4.6. Chronic hemolytic anemia |
| 1.5. Persistent pulmonary hypertension of the newborn |
| 1.5.1. Pulmonary veno-occlusive disease (PVOD) and/or pulmonary capillary hemangiomatosis (PCH) |
| 2. Pulmonary hypertension due to left heart disease |
| 2.1. Systolic dysfunction |
| 2.2. Diastolic dysfunction |
| 2.3. Valvular disease |
| 3. Pulmonary hypertension due to lung diseases and/or hypoxia |
| 3.1. Chronic obstructive pulmonary disease |
| 3.2. Interstitial lung disease |
| 3.3. Other pulmonary diseases with mixed restrictive and obstructive pattern |
| 3.4. Sleep-disordered breathing |
| 3.5. Alveolar hypoventilation disorders |
| 3.6. Chronic exposure to high altitude |
| 3.7. Developmental abnormalities |
| 4. Chronic thromboembolic pulmonary hypertension (CTEPH) |
| 5. PH with unclear multifactorial mechanisms |
| 5.1. Hematologic disorders: myeloproliferative disorders splenectomy. |
| 5.2. Systemic disorders, sarcoidosis, pulmonary Langerhans cell histiocytosis, lymphangioleiomyomatosis, neurofibromatosis, vasculitis |
| 5.3. Metabolic disorders: glycogen storage disease, Gaucher disease, thyroid disorders |
| 5.4. Others: tumoral obstruction, fibrosing mediastinitis, chronic renal failure on dialysis |
In adults, the most common cause of PH is lung disease (eg, chronic obstructive pulmonary disease [COPD]). An estimated 30,000 persons die each year from COPD, many of whom have PH and resulting RV failure as a contributing cause of death. Patients with interstitial lung disease, cystic fibrosis, sleep apnea syndrome, and lung disorders caused by occupational and other exposures also frequently develop PH. In the United States, approximately 200,000 patients die annually from acute pulmonary embolism, often with acute RV failure due to sudden onset of severe PH. PH is also seen in patients with chronic or recurrent pulmonary embolism, regardless of the source of the embolic.
Estimates of the incidence of idiopathic PAH (IPAH; formerly termed PPH) range from 1 to 2 newly diagnosed cases per million people per year. Its prevalence is approximately 0.1% to 0.2% of all patients who have postmortem examinations. The prevalence of pulmonary vascular disease in patients with other illnesses is not known, but it appears that 2% to 4% of patients with portal hypertension and approximately 0.5% with HIV have PAH. The incidence of PAH in patients with systemic sclerosis (which includes limited systemic sclerosis and diffuse systemic sclerosis) ranges from 8% to 12%. PAH has been reported to occur in 23% to 53% of patients with mixed connective tissue diseases and in 1% to 14% of cases of systemic lupus erythematosus, but it is rare in patients with rheumatoid arthritis, Sjögren syndrome, or dermatomyositis.
Pulmonary vascular obstructive disease associated with congenital heart disease, and in particular associated with unrepaired large, unrestrictive congenital systemic to pulmonary shunts (ie, the Eisenmenger syndrome), develops after a period of decreased PVR and increased pulmonary flow. The high rates of pulmonary vascular obstructive disease in uncorrected congenital heart disease (Table 71–4)9 demonstrate that even if all other causes of death could be eliminated, approximately one-third of patients with congenital heart disease would die from pulmonary vascular disease. Why some patients develop irreversible pulmonary vascular obstructive disease in the first year of life and other patients remain “operable” from a pulmonary vascular disease standpoint into the second or third decade of life or later with the same congenital cardiac defect remains unknown.
| Lesion | % | Total No. | No. at Risk |
|---|---|---|---|
| Ventricular septal defect | 30 | 9000 | 3000 |
| Patent ductus arteriosus | 9 | 2700 | 900 |
| Atrial septal defect | 7 | 2100 | 700 |
| Atrioventricular septal defect | 3 | 900 | 800 |
| Aortic stenosis | 5 | 1500 | 0 |
| Pulmonic stenosis | 7 | 2100 | 0 |
| Coarctation | 6 | 1800 | 0 |
| Tetralogy of Fallot | 5 | 1500 | 200 |
| Transposition of the great arteries | 5 | 1500 | 500 |
| Truncus arteriosus | 1 | 300 | 300 |
| Hypoplastic right heart | 2 | 600 | 50 |
| Hypoplastic left heart | 1 | 300 | 0 |
| Double-outlet right ventricle | 0.2 | 60 | 60 |
| Total anomalous pulmonary venous connection | 1 | 300 | 300 |
| Univentricular heart | 0.3 | 90 | 90 |
| Miscellaneous | 17.5 | 5250 | 2625 |
| Total | 100.0 | 30,000 | 9525 (32%) |
Risk factors are categorized based on the strength of the association with PAH and a possible causal role (Table 71–5).
| 1. Definite |
| Aminorex |
| Fenfluramine |
| Dexfenfluramine |
| Toxic rapeseed oil |
| 2. Likely |
| Amphetamines |
| L-tryptophan |
| Methamphetamines |
| 3. Possible |
| Cocaine |
| Phenylpropanolamine |
| St. John’s wort |
| Chemotherapeutic agents |
| Selective serotonin reuptake inhibitors |
| 4. Unlikely |
| Oral contraceptives |
| Estrogen |
| Cigarette smoking |
Familial PAH (FPAH) represents approximately 15% of cases previously considered IPAH. FPAH is inherited as an autosomal-dominant disorder with incomplete penetrance and genetic anticipation.
Mutations in three receptors of the transforming growth factor β family (ie, bone morphogenetic protein receptor 2, activin receptor-like kinase type 1, and endoglin) have been identified in heritable PAH (HPAH), indicating a critical role for transforming growth factor superfamily ligand-receptor interactions in vascular homeostasis and in embryologic development (Fig. 71–1). Exonic mutations in BMPR2 are found in approximately 75% of patients with FPAH and ALK-1 or endoglin mutations in a minority of patients with hereditary hemorrhagic telangiectasia and coexistent PAH. BMPR2 mutations are also seen in approximately 25% of patients who were previously classified as IPAH without a family history of PAH but are now classified under heritable PAH with the subheading sporadic PAH. IPAH is now reserved for patients without a family history and without an identified genetic abnormality. Mutations in BMPR2 confer a 10% to 20% chance of developing PAH in a carrier’s lifetime. Hence gene-gene or gene-environmental interactions appear to be involved to promote or prevent pulmonary vascular disease. BMPR2 mutations have also been identified in other PAH cohorts, including appetite suppressant–induced PAH and PAH associated with congenital systemic to pulmonary shunts. To date, BMPR2 mutations have not been reported in PAH related to connective tissue disease, HIV, or portal hypertension.
Figure 71–1.
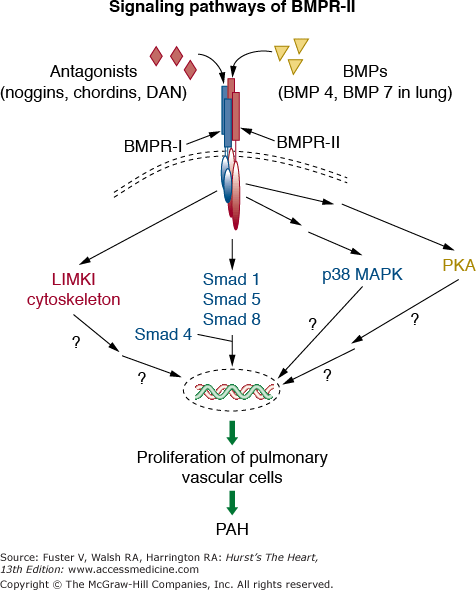
Signaling pathways of the bone morphogenetic protein receptor type II (BMPR II). In the extracellular space, the receptor ligands BMPs bind directly to the BMPR-II on the cell membrane. The bioavailability of BMPs is regulated by the presence of BMPR-II receptor antagonists such as noggins, chordins, and DAN (differential screening-selected gene aberrative in neuroblastoma). The binding of ligands to BMPR-II leads to the recruitment of BMPR-I to form a heteromeric receptor complex at the cell surface. This complex results in the phosphorylation and activation of the kinase domain of BMPR-I. The activated BMPR-I subsequently phosphorylates and activates cytoplasmic signaling proteins Smads (Smad 1, 5, and 8). Phosphorylated Smads bind to the common mediator Smad 4, and the resulting Smad complex moves from the cytoplasm into the nucleus and regulates gene transcription. Other downstream signaling pathways that can be activated after the engagement of BMPR-I and BMPR-II by BMPs include cell-type dependent activation of p38 mitogen-activated protein kinase (p38 MAPK) and protein kinase A (PKA). In addition, the cytoplasmic tail of BMPR-II has been shown to interact with the LIM motif-containing protein kinase 1 (LIMK1) that is localized in the cytoskeleton. Germ-line mutations of the gene encoding BMPR-II underlie heritable pulmonary arterial hypertension (HPAH), which is characterized by the abnormal proliferation of pulmonary vascular cells. However, the specific cytoplasmic proteins and nuclear transcription factors that are involved in the development of HPAH have not been identified.
The gold standard for the confirmation of suspected PH is right heart catheterization. This enables the direct determination of right atrial and ventricular pressures, pulmonary arterial pressure, pulmonary capillary wedge pressure (as an approximation of pulmonary venous pressure), pulmonary blood flow (cardiac output when there are no systemic to pulmonary shunts), and the responses of these parameters to interventions (eg, acute vasodilator testing, oxygen supplementation, rapid intravenous volume loading, or exercise). However, the skilled clinician can often suspect PH on the basis of the assessment of an elevated jugular venous pressure pulsation and a loud P2 (see Chap. 14). PVR is calculated from the measurements and samples obtained during cardiac catheterization (see Table 71–2).
The findings on the chest radiograph depend on the duration of the PH and its etiology (see Chap. 17). The characteristic findings of PH are enlargement of the pulmonary trunk and hilar vessels in association with attenuation (pruning) of the peripheral pulmonary arterial tree (Fig. 71–2). Right-sided heart enlargement can best be detected radiographically on the lateral view as fullness in the retrosternal airspace. In PH due to pulmonary diseases, changes in the lungs (eg, hyperinflation, fibrosis) and in the position of the heart and diaphragm often mask the radiologic changes of PH. Contrast angiography has a role in the workup for PH when chronic thromboembolic disease, which may be treated surgically, is suspected.10
Figure 71–2.
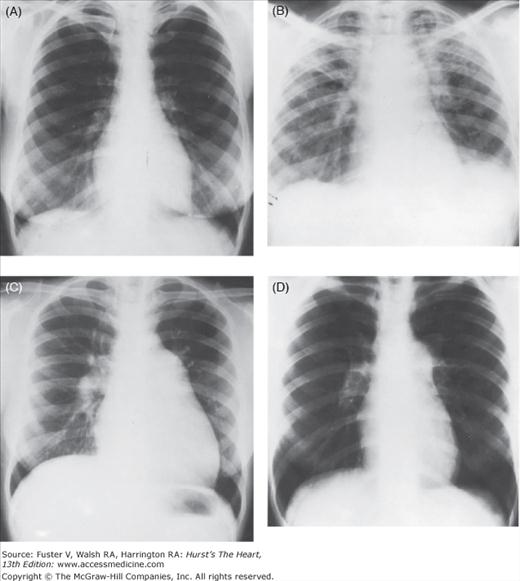
Cardiac silhouette in four patients with severe pulmonary hypertension on admission to the hospital. A,B. Idiopathic pulmonary arterial hypertension (IPAH) showing different stages in the evolution of right-sided heart failure. C. Widespread pulmonary fibrosis. D. Systemic lupus erythematosus proven by lung biopsy. This radiograph is indistinguishable from that of IPAH.
The electrocardiogram (ECG) can disclose hypertrophy of the right ventricle and is more reliable in nonrespiratory etiologies than in obstructive airways disease or parenchymal lung disease (see Chap. 15).
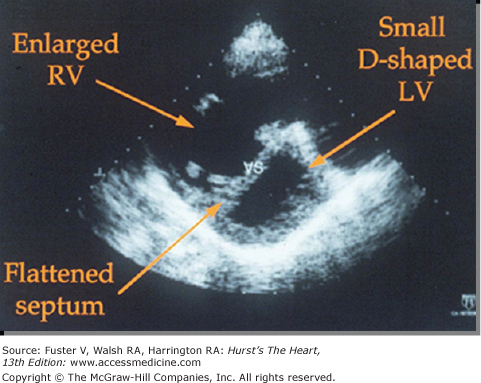
The amount of reliable information obtained by Doppler and two-dimensional echocardiography depends greatly on the commitment of individual clinics to standardizing and perfecting these noninvasive techniques (see Chap. 18). In general, echocardiographic techniques have proved useful in providing a measure of right ventricle thickness as an index of RV hypertension (Fig. 71–3). Estimates of RV systolic pressure (RVSP) can be obtained by determining regurgitant flows across the tricuspid valves using continuous-wave Doppler echocardiography.11 However, echocardiography not infrequently overestimates RVSP; therefore, the diagnosis of PH should not be made without confirmatory right heart catheterization. Echocardiography is an attractive alternative to repeated cardiac catheterization in following the course of the disease and assessing the effects of therapeutic interventions in some patients (see Chap. 18).
Ventilation–perfusion scans are of most value in the diagnosis and exclusion of chronic thromboembolic disease (see Thromboembolic Disease below; also Chap. 72).
The response of the RV to exercise can be assessed by calculating the change in RV ejection fraction (RVEF) with exercise using radionuclide angiography. Scintigraphy using thallium-201 also has been useful in detecting hypertrophy of the right ventricle caused by PH (see Chap. 21). Magnetic resonance imaging has more recently been useful in assessing disease severity (eg, RV mass, RVEF) and effects of therapeutic interventions.
The sampling of lung tissue by open thoracotomy or thoracoscopy is rarely needed to make an accurate diagnosis. There are however exceptions such as confirming suspected pulmonary vasculitis. Furthermore, the procedure carries substantial risk in these hemodynamically compromised individuals. Attempts to predict responsiveness to vasodilators on the basis of lung biopsy have had limited success,12 most likely due to significant heterogeneity in the pulmonary vasculature. Thus the hemodynamic assessment by cardiac catheterization gives a more overall assessment of the degree of pulmonary vascular disease as opposed to an isolated piece of lung tissue. Lung biopsy had been previously considered a necessary component of the diagnostic workup for patients suspected of having pulmonary venoocclusive disease or pulmonary capillary hemangiomatosis; however, both conditions can now be diagnosed by chest CT when performed and interpreted by radiologists who are attuned to these diseases, thereby avoiding an invasive procedure that carries not an insignificant risk.
Pulmonary Hypertension Associated with Cardiac or Pulmonary Disorders
Cardiac and/or respiratory diseases are the most common causes of PH. Cardiac disease leads to PH by increasing pulmonary blood flow (eg, large left-to-right shunts) or by increasing pulmonary venous pressure (eg, LV failure). Invariably, secondary influences such as intimal proliferation in the pulmonary resistance vessels add a component of obstructive pulmonary vascular disease.13 In respiratory disease, the predominant mechanism for the PH is an increase in resistance to pulmonary blood flow arising from perivascular parenchymal changes coupled with pulmonary vasoconstriction and remodeling because of hypoxia. In pulmonary thromboembolic disease, clots in various stages of organization and affecting pulmonary vessels of different size increase resistance to blood flow.14
The mechanisms of PH are often quite different in acquired disorders of the left side of the heart than in those of congenital heart disease.
LV failure is the most common cause of PH. Among the various etiologies, myocardial disorders and lesions of the mitral and aortic valves predominate. Both categories of lesions lead to an increase in pulmonary venous pressure that, in turn, evokes an increase in pulmonary arterial pressure. Presumably, the increase in pulmonary arterial pressure is reflex in origin. In time, three types of morphologic changes supervene: (1) occlusive intimal and medial changes not only in pulmonary venules and veins, but also in the precapillary vessels; (2) perivascular interstitial edema and fibrosis that, under the influence of gravity, cause vascular and perivascular changes to be most marked in the dependent portions of the lungs; and (3) occlusion of small pulmonary vessels by emboli or thrombi in situ when the RV fails and cardiac output decreases. Chapter 28 discusses the medical management of myocardial failure. The treatment of congenital heart disease and of mitral valvular disease is usually mechanical (eg, surgical or interventional such as closure of an atrial septal defect or balloon mitral valvuloplasty). When the PH is due to pulmonary venous hypertension, relief of the pulmonary venous hypertension, as by mitral valve commissurotomy or replacement, will reverse the pulmonary vascular disease in virtually all cases, and even if the PVR does not return to normal, PVR will decrease in virtually all instances to a level that is not clinically significant. An elevated PVR should not be a contraindication to surgery or an interventional procedure to eliminate a discrete site of obstruction causing pulmonary venous hypertension. However, this does not mean that the patient is not without risk for acute pulmonary hypertensive crises, which can be life-threatening, at the time of the intervention. LV failure is the most common cause of RV failure; the level of PH that accompanies LV failure is infrequently sufficient to account for RV failure. RV failure, secondary to LV failure, is usually attributed in part to failure of the muscle in the shared ventricular septum. It turns out, for reasons that remain unclear, that it is the degree of RV hypertension (ie, PH) and the RVEF that are prognostic for LV failure and not the degree of LV failure.
Heart failure is a growing public health problem, with a prevalence of approximately 5 million patients in the United States, and an incidence of more than 555,000 patients.15 Heart failure can be due to abnormalities in systolic and/or diastolic function.16 The incidence of heart failure resulting from abnormalities of diastolic dysfunction with normal systolic function has increased to more than 50% of heart failure patients overall.16-20
Diastolic heart failure, also known as heart failure with preserved ejection fraction, has emerged as a distinct entity, with clear differences in its pathophysiology, diagnostic evaluation, and prognosis compared with heart failure with decreased systolic function.21 The clinical syndrome includes limitations in exercise capacity and fluid retention, with peripheral edema, abdominal ascites, and pulmonary congestion.22 It is largely a clinical diagnosis based on history and physical examination. Patients with heart failure with preserved ejection fraction tend to be older females and have a history of systemic hypertension, LV hypertrophy, obesity, lipid abnormalities, diabetes, myocardial ischemia, coronary artery disease, and/or atrial fibrillation.16,19,20,23 Although the prognosis for these patients was previously considered better than for patients with a decreased LV ejection fraction,20,23-25 recent data suggest otherwise.26 Pulmonary venous hypertension due to diastolic dysfunction27,28 is often more severe than expected. A history of conditions associated with diastolic abnormalities and evidence on diagnostic testing with electrocardiography, chest radiography, computed tomography, and/or echocardiography consistent with left heart disease (eg, left atrial enlargement, LV hypertrophy, or infiltrative disease) should raise suspicion of diastolic heart failure. In some patients LV end-diastolic pressure is normal at rest but increases with exercise. Testing with agents that increase inotropy (eg, dobutamine, rapid intravenous volume loading, or exercise during cardiac catheterization) may identify compliance abnormalities.
There is currently no evidence to support the use of any targeted PAH therapies in patients with diastolic heart failure.
The Eisenmenger syndrome represents an advanced stage of structural and functional changes in the pulmonary vasculature that lead to a progressive increase in PVR.29 Although it is now seldom seen in industrialized nations, the Eisenmenger syndrome is still frequently seen in developing countries where infant cardiac surgery is largely unavailable. Until recently, the absence of specific treatment options for pulmonary vascular disease associated with congenital cardiac defects resulted in varying degrees of therapeutic nihilism, and little structured management was available.
All congenital cardiac defects with an increased pulmonary blood flow have a propensity to develop pulmonary vascular disease. The likelihood and rate of development of vascular disease is determined by a number of variables that include the quantum of left-to-right shunt, the nature of the underlying defect, and duration of exposure of the pulmonary vascular bed to increased flow. Left-to-right shunts at the level of the ventricles or great arteries (post-tricuspid) have a greater chance of developing pulmonary vasculopathy as compared with shunts at the atrial level (pre-tricuspid). Cyanotic cardiac defects with increased pulmonary blood flow (eg, transposition of the great arteries with ventricular septal defect or patent ductus arteriosus, truncus arteriosus) are particularly prone to develop early vasculopathy, often during infancy. Additional variables appear to contribute but are poorly understood: The response of the pulmonary vasculature to a high pulmonary blood flow is not uniform from patient to patient and does not occur in a predictable fashion. There appears to be a spectrum in the development of pulmonary vascular disease, with a subset of patients with high PVR and advanced pulmonary vascular occlusive lesions in early infancy at one end of the spectrum and adults who remain operable with large left-to-right shunts at the other end of the spectrum. Conditions such as trisomy 21, thoracic and spinal skeletal deformities, and lung parenchymal disease are often associated with a higher likelihood of developing pulmonary vascular disease. Increased pulmonary blood flow results in endothelial dysfunction with impaired relaxation and increased vasomotor tone accompanied by histologic changes in the vessel wall. In addition to the signs and symptoms seen with other forms of PAH, patients with the Eisenmenger syndrome have additional comorbid conditions (Table 71–6).
| Abnormalities | Clinical Manifestations |
|---|---|
| Elevated and fixed PVR | Exercise intolerance, dyspnea, syncope, sudden death |
| Secondary erythrocytosis | Hyperviscosity, relative iron, folic acid, and vitamin B12 deficiency |
| Bleeding diathesis | Hemoptysis, cerebral hemorrhage, menorrhagia, epistaxis |
| Right ventricular failure | Liver enlargement, edema |
| Arrhythmias | Syncope, sudden cardiac death |
| Altered red cell rheology and thrombotic diathesis | Cerebrovascular events (stroke or transient ischemic attacks), intrapulmonary thrombosis |
| Renal dysfunction | Increased blood urea nitrogen, hyper-uricemia, and gout |
| Hepatobiliary dysfunction | Calcium bilirubinate gall stones, cholecystitis |
| Infections | Endocarditis, cerebral abscess |
| Skeletal disease | Scoliosis and hypertrophic osteoarthropathy |
Until recently, PAH therapy had been considered potentially harmful in patients with PAH associated with unrepaired congenital heart disease because of the potential to increase right-to-left shunting by reducing systemic vascular resistance to a greater degree than its pulmonary counterpart. However, a number of newer agents now appear promising for patients with Eisenmenger syndrome, including endothelin receptor antagonists and phosphodiesterase-5 inhibitors. The short-term benefits in exercise endurance and hemodynamics may not be predictive of long-term improvement in outcomes; long-term follow-up is needed. With improved intensive care unit and the availability of targeted drugs for PAH (eg, inhaled nitric oxide use in the perioperative period) more patients with what had been previously thought to be inoperable pulmonary vascular disease are now being repaired either surgically or interventionally in the cardiac catheterization laboratory. However, long-term outcome could be affected by eliminating their “pop off” valve with closure of the systemic to pulmonary shunts.
Pulmonary vascular disease is an important component of certain connective tissue diseases, most commonly systemic lupus erythematosus (SLE), the scleroderma spectrum of diseases, and dermatomyositis.30 The lesions may take the form of interstitial inflammation and fibrosis, obliterative disease, or vasculitis, either singly or in combination. Although PH can complicate many connective tissue diseases, it has been documented most often in SLE and progressive systemic sclerosis (scleroderma) and its variant syndromes. The possibility has been raised that IPAH is an inflammatory, or autoimmune, disease. The high frequency of both connective tissue disease and IPAH in women and the occurrence of Raynaud phenomenon in up to 20% of patients with IPAH has been used as additional evidence.31 Additionally, the increased incidence of thyroid abnormalities, and in particular hyperthyroidism, further support the role of autoimmune dysfunction in the pathobiology of PAH. Finally, there is a high incidence of positive serologic tests for antibodies (antinuclear antibody, anti-Ku), particularly in women with IPAH.
The lungs and pleura are frequently involved in SLE, with a reported frequency of up to 70%. Patients with PH and SLE are predominantly women; most of these patients also exhibit Raynaud phenomenon.
The histopathologic lesions in these patients resemble those of IPAH. PH in these patients may originate in thrombi secondary to the hypercoagulable state caused by lupus anticoagulant or anticardiolipin antibodies in the blood. Unfortunately, treatment of PH associated with SLE using either anticoagulants or pulmonary vasodilators has had only modest success. However, patients with active pulmonary vasculitis may either improve or stabilize their vascular disease with immunosuppressive agents.
In progressive systemic sclerosis (which includes limited systemic sclerosis [previously termed the CREST syndrome] and diffuse systemic sclerosis), and in overlap syndromes (eg, mixed connective tissue disease), the incidence of pulmonary vascular disease is estimated to be between 8% and 12%. In these patients, PH is the cause of considerable morbidity and mortality.32 The pulmonary vascular disease may be independent of pulmonary or other visceral disease. As in the case of SLE, the pathology of these lesions is often indistinguishable from that of IPAH. Newer therapies, such as continuous intravenous epoprostenol, endothelin receptor antagonists, and phosphodiesterase type-5 inhibitors, have been shown to improve hemodynamics and exercise endurance33,34 although their impact on survival in this population is less clear.
Thromboembolic disease is a form of occlusive pulmonary vascular disease that may be acute or chronic. In the United States and Europe, clots originating in peripheral veins represent a common cause of chronic occlusive pulmonary vascular disease. Elsewhere in the world, other intravascular particulates may cause pulmonary vascular occlusive disease. For example, in Egypt, where schistosomiasis is endemic, pulmonary vascular disease stemming from ova lodged in pulmonary vessels and hypersensitivity reactions to the organism (usually situated outside the lungs) is common. In some parts of Asia, filariasis is reputed to be an important cause of PH. Tumor emboli to the lungs from extrapulmonary sites (eg, the breast) can cause PH by invading the adjacent minute vessels of the lungs. Intravenous drug use may be associated with talc or cotton fiber embolism to the lungs, which can result in a granulomatous pulmonary arteritis.
The syndromes of thromboembolic pulmonary hypertension can be categorized according to the segments of the pulmonary arterial tree that are primarily affected: (1) small (muscular pulmonary arteries and arterioles), (2) intermediate arteries, and (3) large central arteries. Some overlap is inevitable because clots lodged in large vessels are fragmented by the churning motion of the heart, and both the parent clot and its derivatives tend to move peripherally for final lodging.
At autopsy, small thrombi, predominantly recent in origin, are commonplace in the small pulmonary vessels of patients with PH who have developed heart failure preterminally. In contrast is the syndrome of widespread pulmonary vascular occlusion by organized thrombi in the small pulmonary arteries and arterioles. Once attributed to multiple pulmonary emboli, these lesions are now regarded as organized, in situ thrombi.14 The syndrome is rare and indistinguishable during life from IPAH except by lung biopsy. However, histologic identification of these lesions serves little purpose in management. After a ventilation–perfusion scan has excluded chronic proximal thromboembolism (see below), treatment consists of long-term anticoagulation using warfarin or related agents, antiplatelet agents, or both to prevent further clotting.
This syndrome is by far the most common of the three.14 It is thought to be caused by multiple emboli released from vessels in the upper legs and thighs that progressively amputate the pulmonary arterial tree. Ventilation–perfusion scans and selective angiography demonstrate the pulmonary vascular occlusion, although both studies tend to underestimate the degree of obstruction compared with direct inspection of the vascular tree at surgery or postmortem (see Chap. 72). The major therapeutic concern in these patients is to exclude chronic proximal pulmonary thromboembolism (see below) and to prevent recurrent thromboemboli. Treatment involves the use of anticoagulants of the warfarin type and antiplatelet agents.
In some patients who have survived large to massive pulmonary emboli, resolution fails to occur, and the clots become organized and incorporated into the walls of the major pulmonary arteries, leading to PH (Fig. 71–4). Overwhelming the capacity of the local fibrinolytic mechanisms also allows the clot to propagate, to obstruct large segments of the pulmonary vascular bed, and to decrease the compliance of the central pulmonary vessels. By the time the diagnosis is made, the obstructing lesions in the central pulmonary arteries have become an integral part of the vascular wall through the processes of endothelialization and recanalization.14
Figure 71–4.
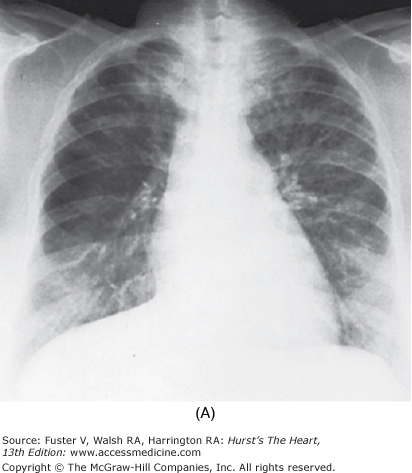
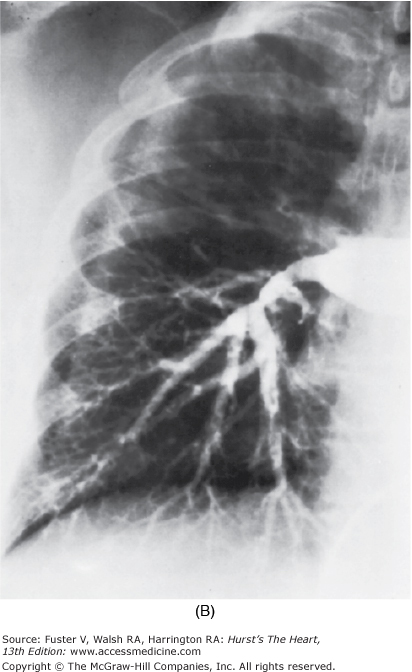
Pulmonary hypertension as a result of an organized clot in central pulmonary arteries. Dramatic relief after pulmonary thromboendarterectomy. A. Chest radiograph. The right upper lobe is strikingly hypoperfused, and the vasculature on the left is quite prominent, reflecting redirection of the pulmonary blood flow to open vessels. B. Angiogram. The flow to the right upper lung is interrupted by the large central clot.
The importance of recognizing proximal pulmonary thromboembolism as a cause of PH is the possibility of relieving the PH by surgical intervention, that is, by pulmonary thromboendarterectomy. Ventilation–perfusion lung scanning is the critical diagnostic test. As a rule, patients with proximal pulmonary thromboembolism show two or more segmental perfusion defects. If the perfusion defects are segmental or larger, selective pulmonary angiography is called for to define the location, extent, and number of pulmonary vascular occlusions.35,36 Although contrast CT may demonstrate chronic thromboembolic PH, it has a 7% false-negative rate. Cardiac catheterization for selective pulmonary angiography also enables hemodynamic assessment. Fiberoptic angioscopy, helical computed tomographic scanning, and magnetic resonance imaging may be helpful in defining the lesions of proximal thromboembolic PH (see Chap. 72).37
Surgery is advocated for patients with PH who have persistent clots in lobar or more proximal pulmonary arteries after at least 6 months of anticoagulation. Thromboendarterectomy is done using a median sternotomy and deep hypothermic cardiopulmonary bypass with intermittent periods of circulatory arrest. Postoperatively, hemodynamic improvement is often quite dramatic.10,36
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


