Pulmonary Hypertension
Srinivas Murali
Introduction
Pulmonary hypertension (PH) is associated with a group of heterogeneous but distinct disorders and is characterized by complex proliferation of the pulmonary vascular endothelium and progressive pulmonary vascular remodeling (1,2). Clinically, PH is defined as a mean pulmonary arterial pressure that is greater than 25 mm Hg at rest or greater than 30 mm Hg during exercise along with a pulmonary capillary wedge pressure or left atrial pressure less than 15 mm Hg, measured at cardiac catheterization. Morphologic evidence for right ventricular remodeling must accompany the hemodynamic derangement (3). This definition was first used in the National Institutes of Health Registry on primary pulmonary hypertension and is now widely accepted by most specialists in the field. Noninvasively, based on echocardiography, PH is defined as a measured Doppler tricuspid regurgitation velocity of greater than 2.8 m/second, which estimates a right ventricular and pulmonary artery systolic pressure of at least 40 mm Hg from the Bernoulli equation (4,5,6,7).
The right ventricle is a low-pressure pump that accommodates well to changes in volume but not pressure (8). The increased afterload to the right ventricle in PH initially results
in the normal adaptive response of hypertrophy and dilation. Eventually, the right ventricular contractile function declines when it is unable to further respond and tolerate this hemodynamic burden. Cor pulmonale is defined as right ventricular dysfunction and failure that directly results from PH associated with chronic parenchymal lung disease (9,10).
in the normal adaptive response of hypertrophy and dilation. Eventually, the right ventricular contractile function declines when it is unable to further respond and tolerate this hemodynamic burden. Cor pulmonale is defined as right ventricular dysfunction and failure that directly results from PH associated with chronic parenchymal lung disease (9,10).
Classification of Pulmonary Hypertension
Even though PH was first described more than 100 years ago, a comprehensive classification for improving clinical understanding was not developed until 1998 (Evian classification). The earlier description of “primary” and “secondary” PH was abandoned and replaced by a classification in an effort to group disorders that share similarities in pathophysiology, clinical presentation, and treatment options (11). It separated conditions that directly affected the pulmonary arterial bed (PAH) from disorders that either involved the pulmonary venous circulation or affected the pulmonary circulation indirectly with involvement of respiratory structure or function. This classification was confusing, however, and had many shortcomings, and did not gain wide acceptance in clinical practice. With improved understanding of the pathogenesis of PAH, a revision to the nomenclature in the classification was proposed in 2003 (Venice classification) not only to reflect current understanding and perspective, but also to permit the widespread use and acceptance of the classification in clinical practice (Table 28.1). The term “primary” pulmonary hypertension (PPH) was replaced by “idiopathic” pulmonary hypertension (IPAH), and, when there was a genetic basis, “familial” pulmonary hypertension. PAH that occurred along with other conditions was referred to as “associated” pulmonary hypertension. This revised classification is accepted by the American College of Chest Physicians (12).
The classification emphasized the role of functional assessment in patients with PH, and a functional classification (WHO Classification), modified from the New York Heart Association functional classification, was adopted to aid clinical definition of the functional limitation (Table 28.2).
Epidemiology of Pulmonary Arterial Hypertension
Idiopathic Pulmonary Arterial Hypertension
The prevalence of idiopathic pulmonary arterial hypertension (IPAH) is not known, and data are somewhat elusive (13,14,15). The prevalence is approximately 1 to 4 new cases per 1 million population. According to World Health Organization (WHO) estimates, there are 5,000 patients with IPAH in the United States and Europe. However, the prevalence may be increasing because of heightened awareness and early recognition in minimally symptomatic patients. According to the National Institutes of Health Registry of 187 patients with primary pulmonary hypertension (PPH) who were followed for 7 years, the mean age at diagnosis was 36 years and the female-to-male preponderance was 2:1 (3). The mean duration of symptoms before diagnosis was 2 years. One-year survival was 68%, 3-year survival was 48%, and 5-year survival was 34%; the median survival was 2.8 years from diagnosis (16). This registry was discontinued, however, before current treatments for PAH
became available. Plans are underway to begin two new registries (funded by industry) that will help to ascertain the prevalence of IPAH in the United States.
became available. Plans are underway to begin two new registries (funded by industry) that will help to ascertain the prevalence of IPAH in the United States.
TABLE 28.1 Revised Venice Nomenclature and classification of Pulmonary Hypertension—2003 | |
|---|---|
|
TABLE 28.2 World Health organization (WHO) Classification of Functional Status in Pulmonary Arterial Hypertension | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
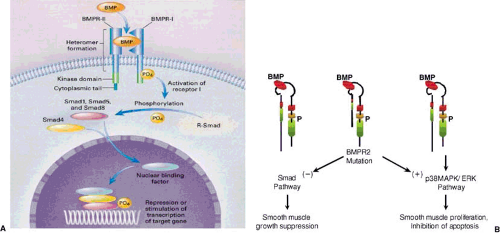 FIGURE 28.1. A: Bone morphogenetic protein (BMP) receptor type 2 (BMPR-2) function. BMPR-1 and BMPR-2 are adjacent on cell membranes. BMP binds to the extracellular domain (ligand binding) of BMPR-2, resulting in the formation of a heteromeric complex with BMPR-1. BMPR-2 then phosphorylates the transmembrane region of BMPR-1, activating the kinase domain. The activated BMPR-1 phosphorylates receptor Smad (R-Smad), thus activating a family of signalling molecules (Smad1, Smad5, and Smad8), which bind with Smad4 and migrate to the nucleus. The phosphorylated Smad complex attaches to a binding factor in the nucleus, and the resulting assembly either stimulates or represses gene transcription by ineracting with DNA. B: Effect of BMPR-2 mutations on intracellular signaling. Normal and abnormal signalling of BMPR-2. Normal pathway: BMPR-2 combines with BMPR-1 and signals through Smad pathway which is not affected by BMPR-2 mutations. This leads to smooth muscle growth suppression. An alternative pathway, through p38 MAP kinase/ERK pathway is the predominant way that mutated BMPR-2 signals leading to smooth muscle proliferation and inhibition of apoptosis. (Adapted from Newman JH, Wheeler L, Lane KB. Mutation in the gene for bone morphogenetic protein receptor II as a cause of primary pulmonary hypertension in a large kindred. N Engl J Med 2001;345:319–324. ) |
Familial Pulmonary Arterial Hypertension
This condition accounts for 6% to 10% of IPAH patients. Mutations in two receptors of the TGF-β superfamily of growth factors have been identified in patients with familial PAH (17,18,19). Greater than 50% of familial cases and nearly 20% of sporadic cases have exonic mutations of the bone morphogenetic protein receptor type 2 (BMPR-2). Bone morphogenetic protein (BMP) is an osteoinductive cytokine that regulates smooth muscle cell growth and apoptosis (Fig. 28.1). It binds to a heteromeric complex of two receptors, BMPR-1 and BMPR-2, phosphorylating a family of signaling molecules (Smad proteins) that translocate to the nucleus and interact with specific proteins messaging suppression of smooth muscle cell growth. In the presence of BMPR-2 mutations, there is disruption of this normal Smad pathway, leading to signaling via an alternate p38 mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) pathway that results in smooth muscle proliferation and inhibition of apoptosis (20,21,22). The entire interaction linking BMPR-2 mutations to the developmentof PAH is incompletely understood. Because the presence of the BMPR-2 mutation confers only a 15% to 20% lifetime risk of developing PAH, a second hit may be required to complete the interaction between this mutation and the development of PAH. Further studies are needed to delineate all the interactions among genes that may regulate the development of PAH and the environment in persons carrying this mutation (23).
Germline mutations of other genes in the TGF-β receptor family, such as the activin-like kinase type-1 (ALK-1) gene and endoglin (ENG) in patients with hereditary hemorrhagic telangiectasia (Osler-Weber-Rendu disease), also confer susceptibility to PAH (24). Autoantibodies to BMPR-2 or ALK-1 may also play a role in the development of PAH associated with connective tissue disease (25). Genetic polymorphisms of the serotonin transporter (5-HTT) have been implicated in the pathogenesis of PH (26,27,28). The long (L) allele is associated with increased 5-HTT transcription, and homozygosityfor the L-allele (LL) leads not only to an increased risk of developing PAH, but also to early onset of the disease in affected individuals. Whether this functional polymorphism confers susceptibility to PAH associated with other disorders is not known. Genetic testing and counseling have been recommended for relatives of patients with familial PAH.
Besides genetic risk factors there are other risk factors associated with the development of PAH (29) (Table 28.3).
Collagen Vascular Diseases
These are associated with both pulmonary involvement and the development of PAH. About 40% of patients with collagen vascular disease have no interstitial lung disease or pulmonary vascular disease. In the remaining 60%, about 19% have isolated PAH, 22% have interstitial lung disease, and
the rest have both. PAH associated with systemic sclerosis or mixed connective tissue disease is present in 37% of patients, equally divided between diffuse and limited (CREST syndrome) disease; in 14% of patients with systemic lupus erythematosus; and in 5% of patients with rheumatoid arthritis (30,31,32,33,34,35). In a community rheumatology practice setting, approximately 13% of patients with collagen vascular disease were found to have undiagnosed PAH. In another series from Canada, 21% of patients with limited systemic sclerosis and 26% with diffuse sclerosis had associated PAH on a screening echocardiogram. The mean duration of systemic sclerosis was 9 years at the time of diagnosis of PAH. It is not known why only some patients with connective tissue disease develop PAH, and there are no biologic or inflammatory markers that can identify susceptible individuals reliably. Complex interactions among genes, environment, and the immune system are perhaps involved in the initial insult to the pulmonary vasculature in these patients. When PAH develops in patients with systemic sclerosis, it portends a very poor prognosis, far worse than in those with and without interstitial lung disease. The 1-year survival in patients with PAH is 50% and the 3-year survival 30%, compared to greater than 90% and greater than 80%, respectively, in patients without pulmonary hypertension (36).
the rest have both. PAH associated with systemic sclerosis or mixed connective tissue disease is present in 37% of patients, equally divided between diffuse and limited (CREST syndrome) disease; in 14% of patients with systemic lupus erythematosus; and in 5% of patients with rheumatoid arthritis (30,31,32,33,34,35). In a community rheumatology practice setting, approximately 13% of patients with collagen vascular disease were found to have undiagnosed PAH. In another series from Canada, 21% of patients with limited systemic sclerosis and 26% with diffuse sclerosis had associated PAH on a screening echocardiogram. The mean duration of systemic sclerosis was 9 years at the time of diagnosis of PAH. It is not known why only some patients with connective tissue disease develop PAH, and there are no biologic or inflammatory markers that can identify susceptible individuals reliably. Complex interactions among genes, environment, and the immune system are perhaps involved in the initial insult to the pulmonary vasculature in these patients. When PAH develops in patients with systemic sclerosis, it portends a very poor prognosis, far worse than in those with and without interstitial lung disease. The 1-year survival in patients with PAH is 50% and the 3-year survival 30%, compared to greater than 90% and greater than 80%, respectively, in patients without pulmonary hypertension (36).
TABLE 28.3 Risk factors for Pulmonary Arterial Hypertension | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||
Patients with systemic sclerosis who are most likely to acquire isolated PAH generally have limited disease (CREST syndrome), anticentromere antibodies, antinucleolar antibodies including U3RNP, B23, Th/To, and U1RNP, disease onset after menopause, and marked decreases in diffusing capacity to carbon monoxide.
Congenital Systemic-Pulmonary Shunts
The prevalence of PAH in patients with congenital systemic-to-pulmonary shunts is not known. Congenital heart anomalies are present in 0.8% of live births, and there are approximately 1 million adult patients with congenital heart disease in the United States. This number is likely to increase further as operative techniques are refined and definitive surgical repairs are performed in childhood. The high pressure and flow through the pulmonary circulation from a left-to-right shunt leads to the development of PAH. The size and duration of the shunt, genetic factors, and vasoactive mediators all predict the development of PAH in congenital heart disease. BMPR-2 mutations are present in 6% of patients, and high circulating levels of endothelin-1 and norepinephrine are associated with the development of PAH. Actuarial survival in PAH associated with congenital heart disease is better than that seen in other conditions. The 1-year survival is 92%, 2-year survival is 89%, 3-year survival is 77%, and 5-year survival is 77% (37,38).
Pulmonary Arterial Hypertension Associated with Liver Disease
Pulmonary-hepatic vascular disorders in advanced liver disease include hepatopulmonary syndrome and portopulmonary hypertension. Hepatopulmonary syndrome is present in 20% of liver disease patients and is characterized by progressive hypoxemia due to an oxygenation defect from intrapulmonary vascular dilations. It does not cause pulmonary hypertension and resolves after liver transplantation. Portopulmonary hypertension is characterized by pulmonary hypertension but not hypoxemia, and is also present in about 20% of advanced liver disease patients. Three hemodynamic subsets are noted: (a) a hyperdynamic circulatory state is common and is associated with moderate increases in pulmonary pressures, a severe increase in cardiac output, a normal pulmonary capillary wedge pressure, and a reduced pulmonary vascular resistance; (b) excess volume subset is also common and is associated with moderate increases in pulmonary pressure and cardiac output, severe increases in pulmonary capillary wedge pressure, and a normal pulmonary vascular resistance; (c) a vasoproliferative state is rare and is associated with severe increases in pulmonary pressures and vascular resistance, a mild decrease in pulmonary capillary wedge pressure, and a moderate increase followed by a decrease in cardiac output. Liver transplantation generally results in resolution of PH, although it can be risky. The vasoproliferative hemodynamic subset in particular is a contraindication to liver transplantation (39,40,41,42,43).
HIV-Associated Pulmonary Arterial Hypertension
There are more than 1 million people with human immunodeficiency virus (HIV) infection in the United States. HIV-associated PAH is seen in 0.5% of patients. Infection with human herpesvirus-8 (HHV-8), which causes Kaposi sarcoma, has been implicated in the pathogenesis of PH associated with HIV infection. The use of highly active antiretroviral therapy may improve PH associated with HIV infection. The actuarial survival is 58% at 1 year, 39% at 2 years, and 21% at 3 years (44,45,46,47,48,49,50).
Pulmonary Arterial Hypertension Associated with Drugs
Drugs and toxins associated with the development of PAH include fenfluramine anorexinogens, aminorex, amphetamines, and cocaine. The use of these drugs for more than 3 months confers a 30-fold higher risk of PAH than in the general population. The development of PAH is thought to be mediated through interactions with the serotonin transporter located in the pulmonary vascular smooth muscle cell. Individuals with higher baseline serotonin expression related to the L allele of the serotonin gene (LL variant) are thought to be susceptible to anorexinogen-induced PAH. Insufficiency of the KV 1.5 channel activity may also predispose to the development of PAH by augmenting the vasoconstrictor response (51,52,53,54).
Pulmonary Arterial Hypertension Associated with Hemoglobinopathies
Other Causes of Pulmonary Hypertension
Chronic Obstructive Lung Disease
Severe chronic obstructive lung disease is frequently associated with PH. It is the most common cause of pulmonary parenchymal disease in the Unites States. According to the Global Initiative for Chronic Obstructive Lung Disease, it is a disease state characterized by progressive, irreversible airflow obstruction and abnormal inflammatory response and fibrosis in the lungs to noxious particles and gases. The progressive airflow limitation is both from bronchiolitis and from loss of elastic recoil from parenchymal destruction, elastolysis, and loss of alveolar attachments. Morphologically, it is characterized by chronic bronchitis, bronchiolitis, and emphysema. There is permanent air space enlargement in emphysema involving the terminal bronchioles and alveoli. It can either be centrilobular, as seen in smokers, or panacinar, as occurs in α1-antitrypsin deficiency. The parenchymal destruction is driven by an imbalance between proteases and antiproteases along with inflammation that predominantly involves CD 8+ T lymphocytes but also neutrophils and macrophages. The proteases that are upregulated include leukocyte elastase, cathepsin G, matrix metalloproteinases, and cysteine proteinases. The counterbalancing antiproteases, including α1antitrypsin, secretory leukocyte protease inhibitor, and tissue inhibitors of metalloproteinases, are decreased. The inflammatory cells also secrete reactive oxygen species, which provokes release of several proinflammatory cytokines, through the activation of intracellular inflammatory pathways. Endothelin-1 (ET-1) expression is also increased in the pulmonary vasculature in chronic obstructive lung disease, particularly during exacerbations (57,58,59).
Pulmonary Hypertension Associated with Interstitial Lung Disease
Interstitial lung disease may be from connective tissue disease, as previously described, or from idiopathic pulmonary fibrosis. Rarely, pulmonary sarcoidosis can cause interstitial lung disease. Depending on the etiology, the prevalence of PH varies in interstitial lung disease. It has been reported in 31% of patients with idiopathic pulmonary fibrosis. When it is present, patients with idiopathic pulmonary fibrosis have lower diffusing capacity for carbon monoxide and are more likely to require supplemental oxygen. Presence of PH results in greater functional limitation, with lower 6-minute walk distance and higher mortality. There is a linear inverse correlation between pulmonary pressures and clinical outcome, with patients with PH having a threefold higher mortality. In idiopathic pulmonary fibrosis, there is repeated lung injury with progressive fibrotic reaction. The levels of the antioxidant glutathione are markedly depleted, and the resulting imbalance between oxidants and antioxidants promotes fibrosis. There is increased endothelin expression, and progression is not related to inflammation (60,61).
Pulmonary Hypertension and Sleep-Disordered Breathing
Obstructive sleep apnea (OSA) is the most common type of sleep-disordered breathing and affects 5% to 15% of the adult population. Its physiologic consequences include systemic hypertension, stroke, coronary disease, heart failure, atrial arrhythmias, and pulmonary hypertension, especially when there is preexisting pulmonary disease. The precise link between OSA and the aforementioned conditions is unknown. Autonomic dysfunction, increased daytime and nocturnal sympathetic activity, endothelial dysfunction, enhanced leukocyte adhesion, increased platelet aggregation, and activation of proinflammatory and prothrombotic factors such as interleukin-6, fibrinogen, and plasminogen activator inhibitor are present in patients with OSA. Patients with OSA and PH are older and heavier and have worse lung function. Apnea-hypopnea index is a poor predictor of PH. Patients with OSA and PH have decreased nitric oxide activity. The apneic spells associated with OSA are associated with hypoxemia and elevations in pulmonary artery pressures, which resolve with restoration of ventilation. The reasons for persistent PH in OSA are not very clear. However, PH in OSA is generally mild and moderately responds to continuous positive-airway-pressure therapy (62,63,64,65).
Chronic Thromboembolic Pulmonary Hypertension
This form of PH develops in only 0.1% to 4% of patients who survive an acute pulmonary embolism. However, its prevalence may be underappreciated. A history of thromboembolic events is present in 60% of patients, and coagulation disorders, most notably a positive lupus anticoagulant titer, is present in 30% of patients. Serotonin transporter gene polymorphism is seen in 30% of patients with chronic thromboembolic PH. The presence of this genetic polymorphism may unfavorably modulate the clinical course of the disease. Appropriate diagnosis of this form of PH is critical because some patients can be cured with surgical thromboendarterectomy (66,67,68,69,70,71).
Pulmonary Hypertension Associated with Left Heart Disease
Chronic left ventricular failure is a common and important cause of PH. In the United States, there are more than 5 million people affected by left ventricular failure, and approximately 550,000 new cases are diagnosed annually. Heart failure affects 10% of the population older than 65 years of age and is the leading cause of hospitalization among adults. Approximately two thirds of heart failures are secondary to diminished left ventricular contractility or systolic dysfunction, and the remainder are due to impaired left ventricular filling or diastolic dysfunction. Coronary artery disease and primary cardiomyopathy are the most common causes of systolic left ventricular failure, whereas hypertension is the leading cause of diastolic failure.
Advanced heart failure accounts for at least 10% of all heart failure (approximately 500,000 patients) and its prevalence is increasing particularly because of emphasis and monitoring of evidence-based medical therapies and reduction in sudden cardiac death due to prophylactic defibrillator implantation. PH may be either mild or moderate, although it can be
severe in up to one third of the patients. It is critical that every heart failure patient with advanced symptoms undergo a thorough evaluation to ascertain the presence and severity of PH (72,73,74,75).
severe in up to one third of the patients. It is critical that every heart failure patient with advanced symptoms undergo a thorough evaluation to ascertain the presence and severity of PH (72,73,74,75).
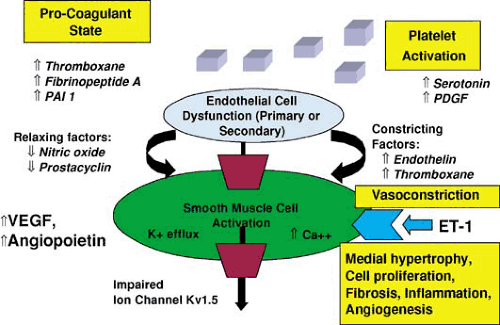 FIGURE 28.2. Pathogenetic mechanisms in pulmonary arterial hypertension. ET-1, endothelin-1; PAI 1, plasminogen activator inhibitor-1; PDGF, platelet-derived growth factor; VEGF, vascular endothelial growth factor. (Adapted and modified from Gaine SP, Rubin LJ. Primary pulmonary hypertension. Lancet 1998;352:719–725. ) |
Pathophysiology of Pulmonary Arterial Hypertension
The pathogenesis of PAH involves several key biologic events (Fig. 28.2). Endothelial cell dysfunction, whether inherited or from other risk factors, results in increased intracellular transcription of constricting factors such as endothelin-1 and thromboxane A2 and decreased activity of relaxing factors like nitric oxide and prostacyclin. This imbalance favors vasoconstriction and signals smooth muscle cell activation and dysfunction, hyperplasia and hypertrophy, inhibition of apoptosis, fibroblast proliferation, collagen deposition, activation of proinflammatory cytokines, and angiogenesis. A number of growth factors including vascular endothelial growth factor and angiopoietin are upregulated, which also promotes cell proliferation and angiogenesis. Impaired function or insufficiency of the voltage-gated potassium-ion channels (KV 1.5) on the pulmonary vascular smooth muscle cell results in efflux of potassium and increased intracellular calcium, which induces further pulmonary vasoconstriction. Platelet activation releases platelet-derived growth factor and serotonin into the circulation. Increased availability of thromboxane A2, fibrinopeptide A, and plasminogen activator inhibitor-1 creates a procoagulant milieu within the pulmonary circulation predisposing to in situ thrombosis. Vasoconstriction, cell proliferation, fibrosis, angiogenesis, and thrombosis combine to produce progressive and deleterious pulmonary vascular remodeling. The list of mediators that are dysregulated in PAH are shown in Table 28.4. Of these, endothelin (ET-1), prostacyclin, and nitric oxide have been studied extensively (76,77).
TABLE 28.4 Current Pharmacologic Treatments for Pulmonary Arterial Hypertension | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||
The Endothelin System
ET-1 is synthesized and released not only by the endothelial cell, but also by other cell types such as leukocytes, macrophages, cardiomyocytes, vascular and airway smooth muscle cells, and mesangial cells. Physicochemical factors such as hypoxemia, pulsatile stretch, low shear stress, and other factors including proinflammatory cytokines, transforming growth factor-β, endotoxin, and a number of circulating neurohormones promote endothelin synthesis and release. Both nitric oxide and prostacyclin inhibit endothelin synthesis. The message for endothelin synthesis from the nucleus results in
the release of an inactive precursor prepro-endothelin into the cytoplasm. This precursor is then cleaved by a furin-like enzyme into an inactive intermediate called big-endothelin. The big-endothelin (big ET-1) is further cleaved by the endothelin-converting enzyme (ECE) to active ET-1 (78,79,80,81,82,83,84).
the release of an inactive precursor prepro-endothelin into the cytoplasm. This precursor is then cleaved by a furin-like enzyme into an inactive intermediate called big-endothelin. The big-endothelin (big ET-1) is further cleaved by the endothelin-converting enzyme (ECE) to active ET-1 (78,79,80,81,82,83,84).
After release, ET-1 mediates its effects through interaction with two classes of cell surface receptors, both of which are G protein–coupled transmembrane proteins, with different molecular and physiologic functions. ET-1 binding to these receptors activates the phosphatidyl inositol phospholipase C pathway, which in turn signals several key cell-specific events. The endothelin type A (ET-A) receptors are located mainly on the vascular smooth muscle cells and mediate vasoconstriction and cell proliferation. They are also present on the cardiac myocyte, cardiac fibroblast, lung, kidney, and brain. The endothelin type B (ET-B) receptors are present predominantly on endothelial cells but are also located on the vascular smooth muscle cells and other organs such as the heart, kidney, brain, intestine, and melanocytes. Activation of these receptors on endothelial cells mediates nitric oxide–dependent vasodilation and prostacyclin release as well as ET-1 clearance particularly in the pulmonary and renal vascular beds, whereas those on the smooth muscle cells mediate vasoconstriction. The net biologic effect of ET-1 depends on the density of ET-A receptors on the vascular smooth muscle cell and the ET-B receptors on the endothelial cell. ET-B receptors are also important for sodium and water absorption in the distal renal tubules.
Under physiologic conditions, ET-1 causes vasoconstriction, cell proliferation, and differentiation determined by a complex interplay between its effects on ET-A and ET-B receptors. ET-1 is important for maintenance of basal tone in many vascular beds. Other effects of ET-1 include a reduction in heart rate, a decrease in coronary blood flow and coronary sinus oxygen saturation, and an increase in cardiac contractility. In pathologic states, the ET receptors are regulated differently, which results in different acute and chronic biologic effects. The acute effects involve vasoconstriction and inflammation, whereas the chronic effects include fibroblast proliferation, synthesis of extracellular matrix components, hypertrophy of cardiac myocytes, neurohormonal secretion, and cell proliferation. ET-1 increases plasma renin activity, enhances conversion of angiotensin I to angiotensin II, and augments the synthesis and release of aldosterone from the adrenal glands. In patients with chronic heart failure, plasma ET-1 levels are increased and correlate directly with functional impairment and inversely with left ventricular ejection fraction. There is a direct correlation between plasma ET-1 levels and left ventricular filling pressures and an indirect correlation with survival and need for transplantation in chronic heart failure patients. Unlike other neurohormones, however, plasma ET-1 also correlates with the severity of pulmonary hypertension in chronic heart failure patients. ET-1 expression in the pulmonary vasculature is increased in patients with PAH. Plasma ET-1 levels are elevated not only in patients with IPAH, but also in patients with pulmonary hypertension associated with connective tissue diseases and congenital heart disease. The high plasma levels of ET-1 in chronic heart failure and pulmonary hypertension may be not only from increased synthesis, but also from reduced clearance in the lung.
Antagonism of the ET system can be achieved either by ECE inhibition or by blocking ET-A and ET-B receptors. Most ECE inhibitors also inhibit neutral endopeptidase (NEP), and so they not only inhibit ET-1 production, but also prevent the metabolism of vasodilating mediators such as atrial natriuretic peptide and bradykinin. The effectiveness of ECE inhibitors may be limited by non-ECE–mediated conversion of big ET-1 to ET-1 (ECE escape). ET-receptor antagonists either selectively block ET-A or ET-B receptor or block both. There are “mixed” or “dual,” nonselective ET receptor antagonists that block both ET-A and ET-B receptors and selective ET-A receptor antagonists that predominantly block ET-A receptors. Bosentan is the prototype nonselective ET-receptor antagonist (20-fold ET-A selective over ET-B), and ambrisentan (100-fold ET-A selective) and sitaxsentan (6,500-fold ET-A selective) are ET-A–selective agents (85,86,87).
Prostacyclin Pathway
Prostacyclin is a major lipid-mediator product of endothelium. It relaxes smooth muscle by increasing intracellular cyclic adenosine monophosphate and inhibits platelet aggregation and smooth muscle cell proliferation. Its production depends on prostacyclin synthase. The expression of prostacyclin synthase is downregulated in the small, medium, and large pulmonary vessels in patients with IPAH. The excretion of urinary metabolites of thromboxane A2 is increased and that of prostacyclin is decreased in patients with IPAH and PAH associated with collagen vascular disease when compared to normal control individuals (88,89,90).
Nitric Oxide Pathway
The expression of endothelial NO synthase (eNOS)is reduced in lungs of patients with pulmonary hypertension, suggesting that reduced bioavailability of NO is important in the pathogenesis. In response to L-arginine, PAH patients have impaired NO production. Exhaled NO and urinary NO metabolites are also reduced in PAH patients. Treatment with endothelin antagonists partially reverses these abnormalities, which suggests that ET-1 may be partly responsible for the decreased eNOS activity in PAH (91,92,93,94,95). Other potential causes for reduced bioavailability of NO in PAH include oxidant stress, increased levels of asymmetric dimethyl arginine (ADMA), which inhibits NOS activity, and decreased L-arginine levels due to increased activity of arginase released from erythrocytes and endothelial cells (96,97,98,99).
NO mediates its biologic effects through an intracellular second messenger called cyclic guanosine monophosphate(cGMP), which is broken down and inactivated by phosphodiesterases (PDEs), especially the isozymes PDE1 and PDE5. Because PDE5 is abundant in the pulmonary vasculature, its inhibition is an attractive therapeutic target in PAH. Impaired activity of soluble guanylate cyclase (sGC) because of oxidation may cause hyporesponsiveness to NO in PAH (100,101).
Pathways Mediating Pulmonary Hypertension in Left Heart Disease
Left ventricular injury leading to structural remodeling and dysfunction is the seminal event in the progression of heart failure. The translation of injury to remodeling depends on the upregulation and downregulation of several neurohormone and cytokine pathways that results in neurohormonal imbalance (102,103,104,105). The renin–angiotensin–aldosterone system, the sympathetic nervous system, and endothelin are the vasoconstrictor systems that are activated, whereas endogenous vasodilator systems, such as nitric oxide and kinins, are deactivated. All of these systems extensively interact with each other, resulting in pulmonary vascular endothelial cell dysfunction. This triggers pulmonary vasoconstriction and vascular remodeling through multiple mechanisms, leading to the development of pulmonary hypertension. The translation from endothelial cell dysfunction to intimal thickening and medial hypertrophy is not well understood, but it involves endothelin-1 and nitric
oxide, both of which play a critical role in the maintenance of vascular tone in health. Plasma endothelin-1 levels vary directly with pulmonary artery pressures and pulmonary vascular resistance and inversely with stroke volume in heart failure patients with PH. Plasma endothelin-1 level is a direct correlate of mortality in heart failure patients. Left ventricular remodeling also results in mitral regurgitation, which causes left atrial hypertension and further triggers pulmonary vascular endothelial dysfunction.
oxide, both of which play a critical role in the maintenance of vascular tone in health. Plasma endothelin-1 levels vary directly with pulmonary artery pressures and pulmonary vascular resistance and inversely with stroke volume in heart failure patients with PH. Plasma endothelin-1 level is a direct correlate of mortality in heart failure patients. Left ventricular remodeling also results in mitral regurgitation, which causes left atrial hypertension and further triggers pulmonary vascular endothelial dysfunction.
Other Pathways
Vasoactive intestinal polypeptide (VIP) is an important neurohormone that is involved in the water and electrolyte secretion in the gut. It inhibits platelet activation and vascular smooth muscle cell proliferation. It is a pulmonary vasodilator, bronchodilator and decreases pulmonary artery pressures and pulmonary vascular resistance in experimental models of PAH. Its biologic action is mediated through activation of two specific VIP receptors (VPAC-1 and VPAC-2), which in turn, promotes cAMP and cGMP signalling. Serum VIP levels are reduced in PAH patients compared to normal subjects and patients with parenchymal lung disease. VIP expression in the tunica media is also reduced in PAH.
Platelet derived growth factor (PDGF) is a mitogen that is up regulated in PAH. PDGF signalling promotes pulmonary vascular smooth muscle cell proliferation in experimental models of pulmonary hypertension. PDGF receptor inhibition has been shown to reverse pulmonary vascular remodeling. Angiopoietin expression has also been shown to be increased in the lung of patients with chronic thrombo–embolic PH. Additionally, matrix metallo-proteinases (MMP) 1 and 9 are also upregulated in the lung tissue. As in chronic heart failure, sympathetic nervous system, renin-angiotensin aldosterone, and B-type natriuretic peptide activity are all increased in patients with PAH.
Clinical Diagnosis of Pulmonary Hypertension
Screening and Early Detection
Patients who have conditions that are associated with PAH should be screened for early recognition (106,107,108). Family members of affected patients or patients who have a history of use of fenfluramine appetite suppressants, current or prior use of amphetamines, or cocaine should also be screened. Accordingly, PAH should be suspected particularly if patients develop unexplained dyspnea. However, many of these patients are either asymptomatic or minimally symptomatic. Although exertional dyspnea is the most frequently encountered symptom, it is sometimes accompanied by exertional fatigue and exercise intolerance. Patients with a history of acute pulmonary embolism should be carefully evaluated for chronic thromboembolic pulmonary hypertension. Similarly, patients with chronic obstructive lung disease, interstitial lung disease, and sleep-disordered breathing should be screened for development of PH if they develop worsening symptoms. Patients with left heart disease who deteriorate on medical therapy or who become refractory to medical therapy should be suspected to have developed associated PH. Transthoracic echocardiogram is the most commonly used screening test for PH. If the resting study is normal, an exercise echocardiogram should be considered to look for exercise-induced PH. The key is to have a high index of suspicion in all patients with the aforementioned conditions (37).
Signs and Symptoms
Although exertional dyspnea and reduced exercise capacity are frequent symptoms, some patients have very nonspecific complaints, such as persistent fatigue, weight gain, and abdominal bloating. Angina and syncope are less common symptoms and frequently portend a poor prognosis. In the later stages, orthopnea, paroxysmal nocturnal dyspnea, and peripheral edema develop indicating the presence of right ventricular failure. Of course, symptoms related to the associated conditions may be present (37).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


