Pulmonary Complications of Intra-abdominal Disease
INTRODUCTION
Intra-abdominal pathology may have distant effects on the pulmonary vascular bed, the pulmonary parenchyma, or the pleural space. This chapter reviews the pulmonary complications of intra-abdominal disease, focusing on the pathology, epidemiology, clinical features, and management. Topics are organized according to the primary abdominal organ system in which the disease is centered.
PULMONARY COMPLICATIONS OF HEPATIC DISEASE
A number of respiratory complications of hepatic disease are well recognized, including portopulmonary hypertension (POPH), hepatopulmonary syndrome (HPS), hepatic hydrothorax (HH), and spontaneous bacterial empyema (SBEM). Each is discussed in the subsequent sections.
 PORTOPULMONARY HYPERTENSION
PORTOPULMONARY HYPERTENSION
POPH is defined as pulmonary hypertension (PH) occurring in the setting of liver disease, or more specifically, in the setting of portal hypertension.
Epidemiology And Pathogenesis
Approximately 2% to 6% of patients with decompensated liver disease develop POPH.1–3 Based on multicenter, case-control studies, autoimmune hepatitis and female gender are identified risk factors for the development of POPH; therefore, the role of hormonal and immunologic influences in the pathogenesis of POPH is a focus of investigation.4,5
Patients with POPH have measurable alterations in levels of pulmonary vasoactive substances. For example, prostacyclin is a potent pulmonary vasodilator that also has antithrombotic and antiproliferative properties; levels of prostacyclin are decreased in the lungs of patients with PH. In patients with POPH, loss of endothelial prostacyclin synthase expression compared with normal controls has been demonstrated.6 Endothelin-1 (ET-1), a pro-proliferative and vasoconstrictive agent implicated in the pathophysiology of other types of PH, has also been shown to be increased in the circulation of patients with POPH.7
Histologically, POPH appears to be very similar to other types of PH. POPH is a precapillary pulmonary arteriopathy involving multiple layers of the vessel; specifically, POPH is characterized by smooth muscle hypertrophy, adventitial proliferation, and endothelial cell proliferation. In addition, the plexiform lesions characteristic of other types of PH have been demonstrated in patients with POPH (Fig. 98-1).8
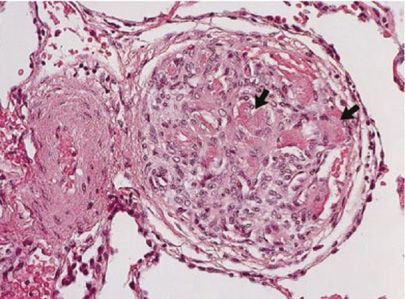
Figure 98-1 Autopsy specimen from a 55-year-old female with cryptogenic cirrhosis and portopulmonary hypertension, showing the plexiform lesion common in pulmonary arterial hypertension. The arrows show acute platelet-fibrin thrombi within the plexiform lesion. (Reproduced with permission from Krowka MJ, Edwards WD. A spectrum of pulmonary vascular pathology in portopulmonary hypertension. Liver Transpl. 2000;6(2):241–242.)
The microvascular changes in POPH result in a pulmonary circulation characterized by increased resistance and low capacitance—the antithesis of the normal pulmonary vascular physiology, which is characterized by a low-pressure and high-flow state. The altered physiology constitutes an increased workload to the right ventricle (RV). Early in the disease process, the RV hypertrophies and increases its stroke volume. Over time, these changes become maladaptive, and the RV dilates and becomes dysfunctional. At this stage, the patient demonstrates progressive right ventricular failure. The clinical course of POPH is similar to that seen in other types of PH (see Chapter 72), justifying its placement in Group 1 of the World Health Organization (WHO) Classification (Table 98-1).9 However, the concomitant presence of end-stage liver disease, with accompanying fluid retention, hypoalbuminemia, coagulopathy, renal dysfunction, and gastrointestinal bleeding, complicates the clinical management of patients.
Source: Adapted with permission from Simonneau G, Robbins IM, Beghetti M, et al. Updated clinical classification of pulmonary hypertension. Journal of the American College of Cardiology. 2009;54(1 Suppl):S43–S54.
Clinical Features
Patients with POPH may present with complaints of fatigue and dyspnea with exertion, which are later accompanied by signs and symptoms of right ventricular failure, such as jugular venous distension, lower extremity edema, presyncope, and syncope.10 The diagnosis can be particularly elusive in the cirrhotic patient, who may have edema from portal hypertension and a spectrum of hemodynamic disturbances. Consequently, the diagnostic workup must be performed carefully to avoid a misdiagnosis of POPH.
The most common hemodynamic derangement in patients with liver disease is not PH, but rather, a state of systemic vasodilation and high cardiac output (CO) induced by splanchnic vasodilation.3 In these patients, hemodynamics are characterized by increased mean pulmonary artery pressure (mPAP), high CO, low pulmonary capillary wedge pressure (PCWP), and reduced pulmonary vascular resistance (PVR). Affected patients do not have small vessel arteriopathy, but rather, increased pressure resulting from high flow through the pulmonary circulation. This hemodynamic pattern is evident in approximately 35% of liver transplant candidates and is not associated with a poor outcome.3
A second abnormal hemodynamic profile present in patients with liver disease reflects volume overload or diastolic dysfunction. With this profile, pulmonary pressures may be elevated, but CO and PCWP are also increased; in addition, PVR is low, indicating pulmonary venous hypertension from elevated left heart filling pressures, rather than small vessel pulmonary arteriopathy.
Finally, a third hemodynamic profile that may be seen in POPH is an elevated pulmonary artery pressure, normal PCWP, high PVR, and high, normal, or low CO—depending on the degree of resultant right ventricular failure and liver disease (Table 98-2).
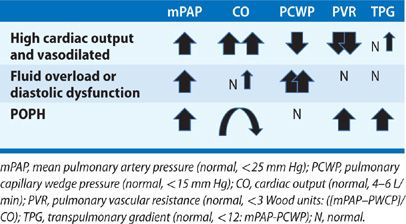
Not infrequently, patients may present with a combination of these hemodynamic profiles, for example, POPH with volume overload. In this case, an increased transpulmonary gradient (TPG) >12 mm Hg (TPG = mPAP – PCWP) may suggest existence of combined volume overload and pulmonary arteriopathy. To characterize such complexity, many have suggested that the hemodynamic definition of POPH be expanded to include mPAP >25, TPG >12, and PVR >3 Wood units.
Diagnosis
To establish a diagnosis of POPH, objective evidence (increased hepatic vein-free pressure to wedge pressure gradient) or clinical evidence of portal hypertension must be present. The diagnosis of POPH is often first suggested by an echocardiogram, but echocardiography is confounded by the previously noted hemodynamic changes seen in cirrhosis.11 Therefore, an echocardiographically estimated peak PA pressure >50 mm Hg or signs of significant right ventricular dysfunction should trigger confirmatory right heart catheterization (RHC). A screening series at the Mayo Clinic determined that this threshold for PA pressure was 100% sensitive in diagnosing POPH, and it minimized unnecessary invasive testing in patients with cirrhosis, who often have a hyperdynamic circulation.3
Clinical Course
Patients with POPH appear to have worse clinical outcomes than patients with other types of PH. The French registry recorded the outcomes of 154 patients with POPH and included both treated and untreated subjects. Five-year survival was reported in 68% of patients. Multivariate analysis identified advanced Childs–Pugh Scores (B and C) and lower cardiac index as independent risk factors for mortality.12
The REVEAL (Registry to Evaluate Early and Long-term PAH Disease Management) registry for PH is an ongoing, multicenter, observational study that has recorded the outcomes of 174 patients with POPH—the largest cohort to date. The data show that patients with POPH have poorer survival and higher all-cause hospitalization rates compared with patients with idiopathic or familial PH, despite the group with POPH having a more favorable initial hemodynamic profile than the others (Fig. 98-2).13 Furthermore, when data were analyzed to derive a risk score for 1-year mortality for all patients presenting with PH, POPH demonstrated a hazard ratio of 3.6 compared to other etiologies.14
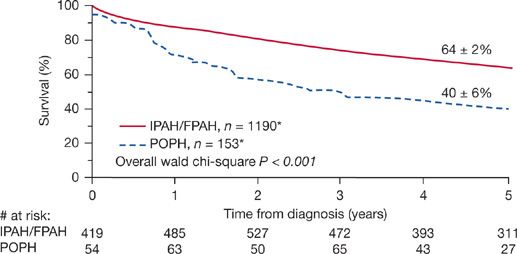
Figure 98-2 Five-year survival from the time of diagnosis of POPH versus IPAH/FPAH. (Data from REVEAL registry. Only patients enrolled within 5 years of diagnosis were included in the 5 years survival from diagnosis curve. Chest. 2012;141(4):905–915.)
Management
POPH is classified as WHO Group I PH, and, therefore, is a treatable category of PH using FDA-approved drugs. However, POPH was excluded from clinical trials assessing use of these agents. Specific data on treatment of POPH are derived from case series and single-center studies, as summarized in the following sections.
Endothelin Antagonists The endothelin antagonists, bosentan and ambrisentan, are established treatments in WHO Group I PAH. Endothelin is a pulmonary vasoconstrictor and pro-proliferative agent, and its antagonism has been shown to improve exercise capacity and hemodynamics in patients with PH. Data on use of these agents specifically in POPH are limited.
Bosentan is a dual endothelin receptor antagonist (ETA and ETB). In small, single-center, uncontrolled, observational trials, patients with POPH treated with bosentan have shown improvements in exercise capacity and survival.15,16 Use of bosentan has been limited in this group due to its well-described hepatotoxicity occurring in a minority of patients (approximately 10%) with PH, but no underlying liver disease. The hepatotoxicity is likely related to the agent’s inhibition of a bile salt transporter; the toxicity is reversible with drug discontinuation.17
Ambrisentan is a selective ETA antagonist and is also FDA approved for the treatment of WHO Group I PAH. Hepatotoxicity has not been reported with this agent. A single-center, uncontrolled, observational trial of ambrisentan in POPH from the Mayo Clinic reported a significant improvement in mPAP and PVR with treatment and no hepatotoxic events.18
Phosphodiesterase Type 5 Inhibitors Two phosphodiesterase type 5 inhibitors, sildenafil and tadalafil, are currently FDA approved for the treatment of WHO Group I PH. The agents vasodilate the pulmonary vascular bed by inhibiting breakdown of cyclic GMP and work within the nitric oxide pathway. Small, uncontrolled, observational trials reveal that sildenafil in POPH increases 6-minute walk test distance and lowers levels of N-terminal prohormone-brain natriuretic peptide, both of which correlate with a better prognosis in patients with PH.19 The reported hemodynamic response has been mixed, with one observational trial reporting an improved PVR in three of five patients at 1 year,20 and another showing improved PVR in all nine patients studied at follow-up, which ranged from 95 to 282 days.21
Prostacyclins Prostacyclin analogs are key agents in the treatment of PAH, and they are considered by many to be the agents of choice for the “sickest” patients. These drugs must be given continuously via subcutaneous or intravenous routes, or intermittently in inhaled form. Current FDA-approved prostacyclins for intravenous use are epoprostenol and treprostinil. Treprostinil is also available for subcutaneous or inhalational administration. In the United States, iloprost is available only in the inhaled form, although it is given intravenously in Europe.
Uncontrolled, single-center series have consistently demonstrated that prostacyclin infusions result in significant improvements in mean PAP, CO, and PVR in patients with POPH.22–24 Furthermore, prostacyclin infusions have been used successfully to improve hemodynamics in patients with POPH in advance of safe pursuit of liver transplantation.24–27 Limited data are available to support the use of inhalational prostacyclins for POPH. Although iloprost has been shown to improve hemodynamics acutely, long-term effects are less certain.28
Liver Transplantation The history of liver transplantation in the setting of POPH is controversial. Early in the history of liver transplantation, the diagnosis of POPH was made in the operating room at the time of the procedure. Patients were neither previously treated for the disease, nor under the care of a medical team familiar with critical care of patients with POPH. Not surprisingly, intraoperative mortality from decompensated right ventricular failure was high.29 Long-term outcome was also adversely affected. Those with severe PH (sPAP >60 mm Hg) had a 9-month posttransplant survival rate of 58%.30 Consequently, severe POPH was considered a contraindication to liver transplantation.
With increasing clinical experience and the advent of new treatments for PH, transplant centers began to report both successful liver transplants in patients with POPH, and, in some, regression of the POPH following surgery.31,32 In 2006, a case series of eight sequential patients with POPH were treated with IV epoprostenol. Of the eight, seven had significant hemodynamic improvement. Six of the seven were listed for liver transplantation, and four of those listed were successfully transplanted. Of those transplanted, survival was 100% at 5 years.26,32 Furthermore, in a retrospective study based on screening of patients with POPH with RHC, 5-year survival for those not transplanted or treated for PH was 15%, whereas 5-year survival for those who were treated for PH and transplanted was 64%.33
Because of the initial poor experiences with POPH patients undergoing liver transplantation, retrospective studies sought to identify risk factors for perioperative mortality. These series have suggested that if a patient has a mean PAP <35 mm Hg and normal right ventricular function, perioperative mortality approaches that of patients without POPH. However, if mean PAP is >50 mm Hg at the time of transplant, mortality approaches 100%.29,31,34
Because there is a role for liver transplantation in selected patients with POPH, the United Network for Organ Sharing (UNOS) allows for upgrade points in the model for end-stage liver disease (MELD) score, facilitating liver transplantation. The MELD system prioritizes liver transplantation for the sickest patients, and the exception system allows extra points for those whose mortality risk is not reflected in their MELD score. Currently, the guidelines indicate that if a patient has a confirmed diagnosis of POPH (using mPAP, PVR, and TPG), and they are treated with pulmonary vasodilator therapy to attain an mPAP <35 and a PVR <5 Wood units, they are eligible for a MELD exception to 22 points, with an increase in their MELD by 10% every 3 months, until they are transplanted.
Liver transplantation in these patients should be performed at a center experienced in pulmonary vascular disease, as the perioperative course may be complicated. Even in patients without POPH, a well-described “reperfusion syndrome” may occur at the time of allograft reperfusion. Reperfusion syndrome may cause acute elevations in PAP and may induce acute, decompensated right ventricular failure in patients with POPH.35,36 The use of intraoperative prostacyclin infusion, inhaled nitric oxide, or intravenous milrinone has been described in this emergent setting; results have been mixed.37,38
Limited data exist regarding the clinical course of POPH after liver transplantation. POPH progression, stability, improvement, and resolution have all been reported. In most of the cases, patients are able to wean from their prostacyclin infusion over a period of months, but some remain on oral pulmonary vasodilators. Available data suggest that 40% to 50% of patients may be able to be weaned from all pulmonary vasodilators, given enough time.24–26
Summary
POPH is an uncommon complication of liver disease, which is characterized by a progressive pulmonary arteriopathy, and which may result in right ventricular failure and death. Patients with POPH appear to have an increased mortality compared with those with similar levels of liver disease or with other types of PH. Specific protocols for optimal use of, pulmonary vasomodulators for POPH are under investigation. However, patients exhibit improved hemodynamics and exercise capacity when receiving therapy for PH. The role of liver transplantation in this setting is arguable, but transplantation may be performed in carefully selected patients and may cure not only their liver disease, but also their POPH.
 HEPATOPULMONARY SYNDROME
HEPATOPULMONARY SYNDROME
HPS is a liver-induced pulmonary vascular disorder characterized by a widened alveolar–arterial oxygen gradient. The widened gradient is the result of intrapulmonary vasodilation (IPVD) in the presence of hepatic disease or portal hypertension. Clinically, IPVD may occur with or without overt hypoxemia, depending on the severity of the disease. HPS increases mortality in patients with liver disease, is without specific therapy, and is completely curable with liver transplantation. Therefore, unlike POPH, the presence of significant HPS is considered an indication for liver transplantation.
Epidemiology and Pathogenesis
The prevalence of HPS is reported as 4% to 32% in cohorts of cirrhotic patients undergoing liver transplant evaluation.39,40 The pathogenesis is poorly understood, but is thought to involve angiogenesis, as well as inadequate synthesis or metabolism by the impaired liver of pulmonary vasoactive substances, such as nitric oxide, prostaglandins, vasoactive intestinal peptide, endothelin, calcitonin, glucagon, substance P, and atrial natriuretic factor.41–43 Nitric oxide has long been implicated in the pathophysiology of HPS, given its known pulmonary vasodilatory effects.44–48 Endothelin has also been implicated in the pathophysiology of HPS. Although endothelin is often thought of as a vasoconstricting agent, and its inhibition is a common therapy for PH, endothelin’s actions vary widely by the receptor to which it attaches. When ET-1 binds to the ETA receptor, the effect is pulmonary vasoconstriction. However, when ET-1 attaches to the ETB receptor, it enhances the activity of endothelial nitric oxide synthase and causes pulmonary vasodilation. In experimental models of HPS, the ETB receptor has been demonstrated to be upregulated; in these animal models, the experimental HPS was reversed by ETB receptor blockade.49,50
Whatever the underlying mechanism of HPS, patients develop IPVD that causes a ventilation–perfusion mismatch and diffusion limitation to oxygenation because of an increase in vascular diameter. The normal pulmonary capillary is 8 to 15 μm in diameter, but in HPS capillaries may dilate to 15 to 100 μm in diameter. Because of this alteration in the normal structure of the alveolar–capillary units, inhaled oxygen does not reach the center of the blood vessel, and some blood returns to the left heart still deoxygenated. In a very rare subset of patients the hypoxemia is the result of true shunt, and AVMs occur with no communication with alveoli.
Clinical Features
Most patients with HPS present with symptoms of chronic liver disease, and a minority (18% in one trial) present with dyspnea as their primary symptom.48 Therefore, a high index of suspicion should be maintained to make a timely diagnosis. Patients may complain of platypnea (dyspnea with standing) or have the physical examination finding of orthodeoxia, which is defined as a PaO2 decrease of 5% or 4 mm Hg upon standing. This combination of findings is attributed to the increases in IPVD in the lung bases, and, therefore, increased ventilation–perfusion mismatch in the standing position. Although the combination of findings is well described in HPS, it is neither common (occurs in 25% of patients) nor pathognomonic; it also has been noted in patients with atrial septal defects, following pneumonectomy, and post-pulmonary emboli.51
On physical examination, patients with HPS may present with spider angiomata, digital clubbing, and peripheral cyanosis.39 The chest radiograph is often normal, but it may reveal bibasilar increased interstitial markings, which may reflect vascular dilation in the bases.52 Pulmonary function testing often reveals reduced diffusion capacity for carbon monoxide, which is out of proportion to other pulmonary function abnormalities.53
HPS has been reported in patients with both acute and chronic liver diseases. Most commonly, it is described in cirrhotics, but HPS has also been documented in the setting of noncirrhotic portal hypertension and acute and chronic hepatitis.48,54 The severity of HPS does not correlate with the severity of the underlying liver disease.
Diagnosis
The diagnosis of HPS requires the presence of (1) cirrhosis or portal hypertension, (2) a widened age-corrected alveolar–arterial oxygen gradient (>15 mm Hg), and (3) demonstration of IVPD on a bubble contrast–enhanced transthoracic echocardiogram (TTE).
Bubble contrast–enhanced TTE is the most sensitive test for the detection of IPVD. However, TTE is a qualitative examination in which saline is agitated and then injected into a peripheral vein. The agitation causes formation of microbubbles that are at least 15 μm in diameter. In normals, the microbubbles are trapped within the pulmonary capillary bed and absorbed. However, if IPVD is present, the bubbles traverse the pulmonary capillaries and appear in the left atrium approximately three to six cardiac cycles after their injection. TTE can also identify an intracardiac shunt, in which case, the bubbles appear in the left atrium within the three cardiac cycles.55
IPVD may also be detected by technetium-labeled macroaggregated albumin lung perfusion scanning (99mTcMAA). In this examination, 99mTcMAA is injected intravenously and uptake detected in the lungs and brain. In the absence of intrapulmonary or intracardiac shunt, the tracer is trapped within the pulmonary circulation and very little is observed on brain imaging. A fractional uptake >5% is considered abnormal. Although MAA scanning allows quantification of the shunt, it cannot differentiate between intracardiac shunt and IPVD. One additional advantage of the 99mTcMAA scan is that it is specific for HPS, even in the presence of intrinsic lung disease. Therefore, it may help to distinguish hypoxemia from HPS from that due to pulmonary parenchymal disease if both are present. 99mTcMAA scanning is less sensitive than TTE for detecting the presence of HPS, and, as demonstrated in several clinical trials, the magnitude of the shunt detected correlates poorly with the degree of hypoxemia.55
Arterial blood gases that quantify PaO2 and the alveolar–arteriolar oxygen gradient for HPS should be obtained with the patient seated at rest and breathing room air. Due to disease-associated alterations in ventilation–perfusion matching with positional changes, along with a propensity for increased IPVD in the lung bases, hypoxemia may worsen with standing and improve in the supine position. Guidelines published in 2004 by the European Respiratory Society Task Force suggest subclassification of HPS according to level of hypoxemia: PaO2 >80 mm Hg is considered mild; PaO2 <80 mm Hg but ≥60 mm Hg is considered moderate; PaO2 <60 mm Hg but ≥50 mm Hg is considered severe, and PaO2 <50 mm Hg is considered very severe.56
Pulse oximetry is a noninvasive screening method for HPS, which may prompt arterial blood gas analysis. A prospective study of pulse oximetry in cirrhotic patients demonstrated that a threshold SpO2 <96% was 100% sensitive and 88% specific in detecting patients with HPS and a PaO2 <70 mm Hg. Application of this threshold cut-point resulted in arterial blood gas testing in 14% of the cohort studied.57
Clinical Course
The presence of significant HPS decreases exercise capacity, impairs quality of life, and increases mortality compared with patients without HPS who have a similar severity of liver disease.58 Furthermore, the majority of patients with HPS progress over time; the average rate of decline in resting PaO2 is 5 mm Hg/y.59 In one multicenter, prospective study, patients with HPS had a doubling in risk of death compared with patients without HPS, despite no differences in rates of listing for liver transplantation, performance of liver transplantation, age, sex, or race.58 A number of centers have implemented screening programs for HPS at the time of liver transplant evaluation. Consequently, a population of patients with IPVD has been identified using screening contrast-enhanced echocardiography without hypoxemia. These patients may not share the poor prognosis of the hypoxemic patients described earlier.60
Management
No medical therapy has been shown to improve patients with HPS. Many agents have been tried unsuccessfully, including norfloxacin, β-blockade, nitric oxide inhibitors, nitric oxide, glucocorticoids, cyclooxygenase inhibitors, indomethacin, somatostatin, cyclophosphamide, and plasma exchange.61 Pentoxifylline, a phosphodiesterase-4 inhibitor that interferes with tumor necrosis factor (TNFα) synthesis, has been demonstrated to have some success in animal models of HPS. In a pilot study of 10 children, 3 months of treatment was associated with a significant increase in PaO2 (>10 mm Hg in all patients); however, the treatment effect disappeared 3 months after drug discontinuation. In addition, the rate of drug discontinuation was 40% due to side effects.62 A pilot study of pentoxifylline in adults showed no significant change in PaO2. The drug was poorly tolerated due to gastrointestinal toxicity.63 Supplemental oxygen is often administered in HPS and may ameliorate the symptoms of hypoxemia.
Liver Transplantation Liver transplantation is the only therapy proved to resolve HPS. Long-term follow-up reveals significant improvement or resolution of HPS in 85% of patients who undergo liver transplantation.64–66 The time required for improvement in oxygenation is variable and may take up to 1 year, or longer.
Hypoxemia due to HPS may complicate the postoperative course in liver transplantation. In one prospective study, the finding of a preoperative PaO2 <50 and an MAA shunt fraction ≥20% was associated with increased postoperative mortality.67 However, other reports suggest that liver transplantation may be safely performed in patients with HPS, including those with severe hypoxemia.68,69
Because of the increased mortality associated with HPS and the favorable outcome for liver transplantation, the UNOS has instituted a MELD exception guideline for affected patients: If the diagnosis of HPS is confirmed with bubble contrast echocardiography and the patient’s PaO2 is <60 mm Hg, an application for MELD upgrade may be submitted. The upgrade increases the patient’s MELD score to 22, regardless of the level of liver disease, with an increase by 10% every 3 months until liver transplantation.70
Summary
HPS is a liver-induced pulmonary vascular disorder characterized by intrapulmonary vascular dilation resulting in ventilation–perfusion mismatch and a diffusion limitation for oxygenation. No known medical treatment for HPS exists, and patients with HPS have a poorer prognosis than for patients with liver disease without HPS. HPS is an indication for, and is curable by, liver transplantation.
 HEPATIC HYDROTHORAX
HEPATIC HYDROTHORAX
HH is defined as pleural fluid (>500 mL) occurring in a patient with liver disease in the absence of cardiac or pulmonary dysfunction. HH may complicate management cirrhosis, and it may contribute to morbidity.
Epidemiology and Pathophysiology
HH occurs in 6% to 10% of patients with end-stage liver disease. Its development is related to anatomic defects in the diaphragm, which allow fluid migration down a pressure gradient from the peritoneum into the negative pressure environment of the pleural space.71,72 It is postulated that as ascites develops, intra-abdominal pressure increases and strains diaphragmatic tissue, which then weakens. Eventually, pleuroperitoneal blebs form and then rupture, creating a communication between the abdominal and pleural cavities. In general, these blebs are <1 cm in diameter and are more common in the right hemidiaphragm, perhaps as a consequence of the more muscular construct of the left hemidiaphragm.73 Although HH usually occurs in the setting of ascites, HH may be present even when clinically apparent ascites is absent, making the diagnosis more challenging.74,75
Clinical Features
HH is right sided in 85% of patients, left sided in 13%, and bilateral in 2%.75 Pleural fluid analysis generally reveals characteristics consistent with ascitic fluid, with slight alterations due to the increased ability of the pleura to absorb free water. HH is transudative, with a total protein level <2.5 g/dL, lactate dehydrogenase fluid-to-serum ratio <0.5, and a total protein fluid-to-serum ratio <2.5. The serum-to-fluid albumin gradient is generally >1.1 g/dL.71,75,76 Occasionally, patients present with hepatic chylothorax. In these cases, the fluid is also transudative, but the fluid-to-serum triglyceride level exceeds 1; in addition, the fluid-to-serum cholesterol ratio is high. Hepatic chylothorax is thought to arise from chylous ascites entering the pleural space through the previously described mechanism.77
Diagnosis
Diagnostic thoracentesis should be performed in the setting of hepatocellular carcinoma, fever, or pain. A prospective series investigating the clinical utility and risks of thoracentesis in the cirrhotic population demonstrated that 30% of patients have an alternative diagnosis to uncomplicated HH. The most common alternative diagnoses are SBEM (see below), pleural tuberculosis, or adenocarcinoma.66 In another large prospective series, thoracentesis was safely performed in cirrhotic patients with mild coagulopathy (PT or PTT ≤ 2× normal, platelet count of 50–99,000[per μL]) without prophylactic plasma or platelet transfusion. Hence, the presence of a mild coagulopathy should not delay diagnostic evaluation.78
 SPONTANEOUS BACTERIAL EMPYEMA
SPONTANEOUS BACTERIAL EMPYEMA
SBEM occurs when a previously existing HH becomes infected. The empyema portion of this term is a misnomer, because diagnosis does not require purulent material in the pleural cavity, but rather, refers to an infection diagnosed by pleural fluid cell count analysis. The diagnosis is made if the pleural fluid has a positive culture and the polymorphonuclear neutrophil (PMN) count is >250 cells/mm3, or, if the culture is negative, the fluid contains >500 PMNs/mm3. The sensitivity of the culture increases from 33% to 77% if blood culture bottles are directly inoculated at the bedside during the procedure.79 The incidence of SBEM has been reported as 13% in hospitalized patients with cirrhosis and a pleural effusion. Simultaneous SBP is a risk factor for the development of SBEM; however, up to 40% of cases are not associated with SBP.79,80 Mortality has been reported as high as 20%. Therefore, clinicians should maintain a high index of suspicion to ensure the diagnosis is made in a timely manner.81
The most commonly identified bacterial pathogens in SBEM are Escherichia coli, Klebsiella pneumoniae, Streptococcus species, and Enterococcus species. Third-generation cephalosporins are the empiric treatment of choice until culture data are obtained.82 Chest tube insertion is not recommended unless frank pus is present in the pleural cavity, given the risks of complications from the procedure in this patient population.
Management
Medical and surgical interventions are employed in the management of HH. Each is described briefly in the subsequent sections.
Diuretics As is the case in management of ascites, the mainstay of treatment of HH is the establishment of negative sodium balance. Current guidelines for management of ascites call for a diet that entails 2 g/d sodium restriction, along with the administration of furosemide and spironolactone in a ratio of 40 mg of furosemide to 100 mg of spironolactone daily. Despite aggressive medical management, 20% to 26% of cirrhotics with ascites are refractory to medical treatment.83
Thoracentesis Thoracentesis can be performed safely in patients with cirrhosis, even in the face of mild coagulopathy. Therefore, therapeutic thoracentesis may be performed to relieve dyspnea in patients with HH that is refractory to dietary sodium restriction and diuretics. Traditionally, removal of no more than 2 L of fluid is recommended at one time because of the risk of reexpansion pulmonary edema and hypotension. However, whether the risks of large-volume thoracenteses apply to this population of patients is unclear.84 Large-volume thoracenteses have been performed safely with close patient monitoring and measurement of end-expiratory pleural pressure. The recommended goal for end-expiratory pleural pressure is >-20 cm H2O.85 Data in support of use of intravenous infusion of albumin to avoid hypotension during thoracentesis are lacking.
Large-volume paracentesis may also be considered for patients in respiratory distress in patients due to ascites or HH. Paracentesis improves dyspnea and lung function within 2 hours of fluid removal. The procedure may provide symptomatic relief for patients with HH and a lower risk than thoracentesis.86
Tube Thoracostomy Although tube thoracostomy may seem an attractive option in patients with recurrent HH, multiple reports have confirmed a high complication rate. Specifically, pneumothorax, hemothorax, empyema, electrolyte abnormalities, and hepatorenal syndrome have all been reported. In one series of 59 patients with cirrhosis and chest tube placement, a complication rate of 80% and mortality rate of 27% were reported. Therefore, this intervention should be avoided.87
Transjugular Intrahepatic Portosystemic Shunt Transjugular intrahepatic portosystemic shunt (TIPS) reduces portal pressure by creating a shunt between the portal and hepatic veins. TIPS placement is the treatment of choice for a refractory HH. In a series of 73 patients with refractory HH who underwent TIPS placement, 79% had a complete or partial response to the procedure; long-term survival rates were consistent with the severity of liver disease.88
TIPS placement is not appropriate for all patients, despite its success in patients with HH and ascites. Patients with severely decompensated liver disease may decompensate further following the procedure because of the shunt’s negative effect on hepatic blood flow. In addition, shunting blood from the portal to hepatic vein increases right atrial and pulmonary artery pressure and may further compromise patients with right-sided heart failure or PH.89 Therefore, MELD >18, PH, large portal vein thrombus, significant hepatic encephalopathy, advanced age (>70 years), or right-sided heart dysfunction are considered relative contraindications for TIPS placement.90 In a high-risk patient, the option of liver transplantation should be discussed before proceeding with TIPS, since transplantation can be used as a “safety net” for clinical decompensation. In a carefully selected population with refractory HH, TIPS is the standard of care.
Surgical Intervention Small case series have documented success in treating HH using open thoracotomy or video-assisted thoracoscopy (VATS).91–94 The procedures are based on identification of diaphragmatic defects and their closure with subsequent pleurodesis. Surgical pleurodesis has a documented success rate of 73% to 100% in preventing recurrence of HH; however, high complication rates have been noted. In one series of 18 patients, a success rate of 48% was noted, but 3-month mortality was 38.9%.95 Therefore, although VATS with pleurodesis may be an option for patients with refractory HH, larger trials are needed to assess safety before this procedure is considered a standard recommendation.
Liver Transplantation Liver transplantation is curative for HH and should be the primary intervention for patients with refractory disease. The presence of HH does not affect ventilator days in the immediate postoperative period, nor does it increase long- or short-term posttransplant mortality.96
Summary
HH may complicate the clinical course of a patient with cirrhosis. A high index of suspicion is needed to diagnose SBEM and to avoid the high mortality rate associated with this condition. Dietary sodium restriction and diuretic use are the mainstays of therapy for pleural effusion, but refractory patients may require therapeutic thoracentesis or TIPS for control. Ultimately, liver transplantation is curative for refractory HH.
PULMONARY COMPLICATIONS OF GASTROINTESTINAL DISEASE
In addition to the aforementioned pulmonary complications of liver disease, diseases of other intra-abdominal organs may have significant effects on the lungs. These include disorders of the esophagus, bowel, and pancreas. Pulmonary complications of renal disease are discussed separately, below.
 GASTROESOPHAGEAL REFLUX DISEASE
GASTROESOPHAGEAL REFLUX DISEASE
Gastroesophageal reflux disease (GERD) affects up to 20% of the population.97 GERD is characterized by reflux of gastric contents into the esophagus due to failure of the protective mechanisms of the upper gastrointestinal tract (see below) and produces symptoms of mucosal damage. In addition to the common symptom of “heartburn,” reflux may be associated with a range of extraesophageal symptoms, including chronic cough, asthma, IPF, bronchiectasis, aspiration pneumonia, and COPD.98
Pathophysiology
Normally, several antireflux mechanisms protect the esophagus and airways from gastric contents: (1) Esophageal peristalsis allows clearance of regurgitated materials; dysmotility is associated with GERD. Abnormal peristalsis is reported in 40% to 50% of affected patients.99 (2) The lower esophageal sphincter (LES) creates a barrier between the esophagus and the stomach. An incompetent LES is associated with reflux. (3) The diaphragm helps prevent reflux through external compression of the lower esophagus. Maintenance of a low thoracoabdominal pressure gradient may be important in minimizing reflux. Obese patients have higher incidence of GERD, which may be a reflection of the increase in intra-abdominal pressure that occurs with obesity.100,101
GERD-related pulmonary disease occurs with gross aspiration of large volumes of gastric contents, recurrent microaspiration, or, possibly, a GERD-induced reflex that causes an increase in vagal tone and resultant bronchospasm.102–104
Diagnosis
Diagnostic testing includes ambulatory esophageal pH monitoring and esophageal impedance testing.
Ambulatory Esophageal pH Monitoring Twenty-four hour ambulatory pH monitoring is considered the “gold-standard” diagnostic maneuver.105 Until recently, this procedure required a catheter-based system, inserted through the nose and placed near the squamocolumnar junction in the esophagus. Various symptom scoring techniques allow linking of patient symptoms and reflux events to determine an association between the two and potential therapeutic intervention. However, the catheters employed are uncomfortable and external, and data suggest that the catheter, itself, may alter patient activity enough to impede the diagnostic accuracy. Recently, a capsule-based system with wireless data transmission has been developed, allowing increased patient comfort and incremental data collection to aid in the diagnosis of GERD. Currently, the American College of Gastroenterology recommends ambulatory pH monitoring for patients with negative findings on esophagogastroduodenoscopy (EGD) who are being considered for an antireflux procedure, and for those failing therapy with a proton pump inhibitor (PPI). Symptom correlation during the procedure is recommended, although its diagnostic role is unproven for extraesophageal symptoms other than chest pain.
Esophageal Impedance Testing Esophageal impedance testing measures changes in tissue resistance to electrical current generated in adjacent electrodes encompassed within an esophageal catheter assembly. The setup allows for identification of retrograde or anterograde food bolus movement along the esophagus. It does not differentiate between acidic, weakly acidic, or alkaline material and, therefore, the technique is often combined with pH monitoring. Impedance testing is more sensitive than pH monitoring alone for detecting the presence of GERD, although it has not been validated for the evaluation of extraesophageal manifestations of GERD.
Clinical Features
Several potential pulmonary associations of underlying GERD should be considered, including asthma, IPF, and chronic cough.
GERD and Asthma The nature of the relationship between GERD and asthma is a subject of controversy, but GERD is present in 15% to 83% of asthmatics.106–108 Several mechanisms by which acid reflux may be related to bronchoconstriction have been described in the literature. Acid reflux into the distal esophagus is postulated to cause bronchoconstriction through vagal stimulation.109,110
Declines in esophageal and tracheal pH coincide with reduced airflow, suggesting that microaspiration of gastric acid may lead to bronchoconstriction.111 Interestingly, some studies have actually suggested the converse—that asthma exacerbations promote GERD by increasing the pleural-peritoneal pressure gradient by lowering pleural pressure,112 by promoting LES relaxation,113 or, perhaps, by a direct effect of bronchodilators on the LES.114–116
Asthma guidelines suggest that GERD be treated in asthmatics who have complaints of heartburn or frequent night-time symptoms. However, in a study of over 400 patients with poorly controlled asthma who received a PPI, treatment had no effect on respiratory exacerbations compared with those who received placebo.117 Current NIH guidelines suggest that treatment of GERD may improve asthma control, citing grade B evidence.118
GERD and Idiopathic Pulmonary Fibrosis Recently, a clinical association has been noted between GERD and IPF; the estimated prevalence of GERD in the IPF population is 90%. This association coincides with a recent shift in thought regarding the etiology of IPF, away from one of active inflammation, and toward one of alveolar injury and maladaptive repair, resulting in fibroblast proliferation and, ultimately, lung fibrosis.119 A strong association between IPF and GERD has been documented in recent clinical trials.120–122 In addition, one retrospective review of 14 patients awaiting lung transplantation for IPF-reported stabilization, but not improvement, of lung function with laparoscopic Nissen fundoplication.123 The mechanistic relationship between IPF and GERD, the best diagnostic modality for assessment, and the optimal treatment algorithm are areas of active investigation. No established guidelines have yet been created.
GERD and Chronic Cough GERD is a well-known cause of chronic cough, and treatment of GERD is considered a mainstay in initial therapy for chronic cough. GERD was identified as a cause of chronic cough in 1981, and since then data have emerged ranking it as the second most common cause.124,125 Twenty-eight percent of patients with chronic cough that improves with GERD therapy deny typical symptoms of GERD.126 Although 24-hour esophageal pH monitoring is considered the most sensitive test for GERD in chronic cough, it is expensive and has limitations. Therefore, the most recent American College of Chest Physicians guidelines on the treatment of cough recommend empiric therapy for GERD in patients whose cough is plausibly related to reflux.127
Treatment
The three main treatments for GERD are antacids, histamine-2 receptor antagonists (H2RA), and PPIs. PPIs have established efficacy over other options and are now the mainstay of therapy. Surgical therapy using Nissen fundoplication is reserved for refractory cases; it may be performed through an open incision or laparoscopically. Finally, new, less invasive procedures to improve the function of the LES are under investigation.
Summary
GERD is a common condition, affecting up to 20% of the population. The reflux of gastric contents into the esophagus may exacerbate pulmonary conditions such as IPF, asthma, and COPD, but neither a causal relationship nor a significant response to therapy has been documented in any of these conditions. GERD is a frequent cause of chronic cough; treatment with a PPI may ameliorate symptoms in affected patients.
 INFLAMMATORY BOWEL DISEASE
INFLAMMATORY BOWEL DISEASE
Pulmonary complications associated with inflammatory bowel disease (IBD) were first described in 1976 in a small series of patients who had both IBD and chronic bronchitis.128 Since that time, a connection between IBD and pulmonary disease has been increasingly recognized, but the pathophysiology of the relationship remains unclear. Both Crohn’s disease and ulcerative colitis (UC) have been associated with a variety of respiratory problems: airway disease, parenchymal lung disease, serositis, and pulmonary vascular disease.
Airway involvement, the most common pulmonary disorder in IBD, may present as bronchiectasis, chronic bronchitis, tracheitis, subglottic stenosis, or, rarely, small airway disease.129–131 Pulmonary parenchymal involvement is less common, but cryptogenic organizing pneumonia, eosinophilic pneumonia, nonspecific interstitial pneumonitis, necrobiotic nodules, and sarcoidosis have been described in IBD.129,132,133 In addition, an exudative pleuritis that does not relate to IBD disease activity has been reported.129 The association between the gastrointestinal and pulmonary “inflammation” is unclear, but the majority of these pulmonary complications appear to be steroid-responsive.132 Finally, patients with IBD have a higher incidence of thromboembolic events than do age-matched controls; the risk of pulmonary embolism does not appear to be related to disease activity.134
PULMONARY COMPLICATIONS OF ACUTE PANCREATITIS
Acute pancreatitis (AP) is an acute inflammation of the pancreas, which may cause both systemic inflammatory response syndrome (SIRS) and multisystem organ dysfunction syndrome (MODS) (see Chapter 142). When MODS accompanies pancreatitis, mortality is reported at 15% to 20%.135,136 Pulmonary complications of pancreatitis are reported in 75% of cases. The most common presentations are hypoxemia without radiologic abnormalities, pleural effusions or, in the sickest patients, acute respiratory distress syndrome (ARDS).137,138
 HYPOXEMIA
HYPOXEMIA
Hypoxemia occurring early in the course of pancreatitis, even prior to development of radiological abnormalities, is well documented and a risk factor for ARDS and increased mortality.137,139,140 Consequently, hypoxemia is included in the pancreatitis severity scores of both Ranson and Imrie.141,142
Several inflammatory mechanisms are implicated in development of the early lung injury of pancreatitis. Increased permeability at the microvasculature leads to alveolar filling and decreased lung compliance. In addition, nitric oxide–related endothelial cell damage occurs, with leukocyte activation and adhesion at the level of the pulmonary microvasculature. Furthermore, the systemic release of pancreatic elastase may contribute to inflammation in the lung parenchyma, and fibrinogen metabolism may allow fibrin deposition in alveoli. Collectively, these processes transform the relatively benign environment of the alveolus into an inflammatory milieu and profoundly affect gas exchange. Early hypoxemia is an independent predictor of poor outcome in AP.140
 PLEURAL EFFUSION
PLEURAL EFFUSION
The presence of a pleural effusion is a marker of the severity of pancreatitis and occurs in 84% of patients with severe disease, but in only 8.6% of patients with mild disease.143 Approximately two-thirds of the effusions are left sided, but they may be bilateral or right sided.144 The effusion is usually small and may be hemorrhagic. Analysis of pleural fluid reveals a very high amylase and elevations in protein and LDH levels when compared with serum.145 Pleural effusions in AP may be caused by transdiaphragmatic lymphatic blockage or arise as a sympathetic effusion due to associated inflammation. A rare complication is a pancreaticopleural fistula, in which the pancreatic duct or a pseudocyst empties posteriorly into the retroperitoneum, tracking up the mediastinum and rupturing into the pleural cavity. The resulting effusions are generally massive and require evacuation and correction of the anatomic defect.146,147 When less than massive, the effusions are treated conservatively and typically resolve with resolution of the pancreatitis.
 ACUTE RESPIRATORY DISTRESS SYNDROME
ACUTE RESPIRATORY DISTRESS SYNDROME
ARDS develops in approximately 15% to 20% of patients with AP. When present, mortality rises >50%.148 The pathophysiologic basis of ARDS in AP is poorly understood, but inflammatory cytokines have been implicated (see Chapter 140). Specifically, trypsin injures the pulmonary vasculature and promotes leukostasis and increased vascular permeability.149,150 Phospholipase A2 (PLA2), which removes fatty acids from phospholipids, may act on surfactant, contributing to respiratory failure.151,152 Multiple other mediators have been implicated, including IL-6, IL-8, NO, TNFα, and substance P. Whatever the underlying mechanism, ARDS is the basis for 50% to 90% of deaths in AP.153 Treatment is supportive and incorporates an alveolar protective ventilator strategy (see Chapter 141).
Summary
AP may cause a SIRS and MODS. One of the early signs of ensuing MODS may be hypoxemia, which is a risk factor for the later development of ARDS and early mortality. Pancreatitis is often accompanied by a pleural effusion, which is exudative and characterized by a high amylase level. In general, these effusions may be handled conservatively. ARDS occurs in 15% to 20% of patients with pancreatitis and is the cause of death in most who do not survive the bout of pancreatitis.
PULMONARY COMPLICATIONS OF RENAL FAILURE
Several notable associations exist between respiratory disease and renal failure, including PH and metastatic pulmonary calcification.
 PULMONARY HYPERTENSION AND RENAL FAILURE
PULMONARY HYPERTENSION AND RENAL FAILURE
PH associated with chronic kidney disease (CKD) is encompassed within Group 5 of the WHO classification of PH (PH with unclear or multifactorial causes).154 CKD is defined by a reduced glomerular filtration rate (GFR) <60/mL/min/1/73 m2) for ≥3 months. Screening for PH in CKD is accomplished using TTE and estimation of right ventricular systolic pressure (RVSP). Generally, RVSP >35 mm Hg is considered “abnormal” in patients with CKD.154 Subsequent to screening, accurate characterization and definitive diagnosis of PH in CKD requires RHC. Patients with CKD are complex with respect to the many factors, acting individually or in combination, that may increase RVSP (Table 98-3).155,156



