CHAPTER 118 Pulmonary Atresia with Intact Ventricular Septum
Pulmonary atresia with intact ventricular septum (PA/IVS) is a rare congenital heart defect occurring at a rate of 4 to 10 per 100,000 live births.1,2 This malformation is characterized by a variably sized right ventricle that has no exit, failing to provide pulmonary blood flow and unable to decompress itself through the interventricular septum. Blood flow through the ductus arteriosus permits survival at birth, but within hours hypoxemia progresses to death as the ductus closes. Thus, without early diagnosis and treatment, PA/IVS is uniformly fatal. Before 1970, reported survival to 3 years of age was less than 3%.3 By the early 1990s, improvements in diagnosis and management resulted in a 3-year survival rate greater than 60%.4 More recent reports indicate that careful initial evaluation and selective management of these patients can achieve survival rates in excess of 90%.5
Because this anomaly is rare and its morphology is heterogeneous, the majority of reports and recommendations have been derived from small series with variable morphologic compositions. Although a great deal has been learned from the experiences of individual centers,6–9 much more has been learned from a prospective multi-institutional study initiated by the Congenital Heart Surgeons Society in 19874,10 and from more recent, population-based studies from the United Kingdom and Ireland1,11 and from Sweden.2 Data acquired from these studies have provided important new insights into the spectrum of morphology, and about outcomes of surgical treatment of this malformation.
Pulmonary atresia with intact ventricular septum has been described by other monikers in the past, including pulmonary atresia with normal aortic root and hypoplasia of the right heart. The first description of pulmonary atresia with an intact ventricular septum was in 1784 by Hunter.12 Connections between the right ventricle and the coronary arteries were described by Grant in 1926 after examining the heart of a 14-month-old girl.12,12a Lauer and colleagues in 1964 were the first to angiographically demonstrate intramyocardial sinusoids.12 Freedom and colleagues postulated in 1974 that the connections between the right ventricle and the coronary arteries could possibly be related to myocardial ischemia.12 The first successful surgical repair of a patient with PA/IVS involved a transventricular valvotomy and was reported by Weinberg and associates in 1962.13
ANATOMY
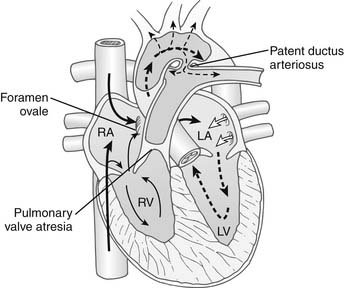
Characteristically, PA/IVS occurs in hearts with situs solitus and atrioventricular and ventriculoarterial concordance. The aortic arch is usually left-sided, and a left-sided ductus arteriosus is typical. The essential feature of this lesion is an absent communication between the right ventricle and the pulmonary trunk (Fig. 118-1). The character of the atretic segment ranges from a thin imperforate membrane to a long section of infundibular muscle without a definable lumen. In contrast to pulmonary atresia with ventricular septal defect, the pulmonary trunk and branch pulmonary arteries are usually near normal in size and configuration. The size and morphology of the right ventricle varies significantly in this condition, and in most patients, the right ventricular cavity is reduced in size. There is a continuum from tiny “unipartite” chambers that have only an inlet component, to larger-than-normal “tripartite” ventricles that have well-defined inlet, trabecular, and infundibular portions. The rare patients with enlarged right ventricular cavities also may have Ebstein’s anomaly with severe tricuspid regurgitation. More typically, the cavity is small, and marked hypertrophy of the right ventricular wall is present, often contributing to obliteration of the outflow (infundibular) portion of the cavity. The tricuspid valve is usually small with thickened leaflets and abnormal chordae. The diameter of the tricuspid valve correlates with the size of the right ventricular cavity and provides a useful index of right ventricular size. The right atrium is enlarged, and an interatrial communication, usually a patent foramen ovale, is present. At birth, the ductus arteriosus is patent, providing the only blood flow to the lungs. Significant aortopulmonary collateral arteries are uncommon.
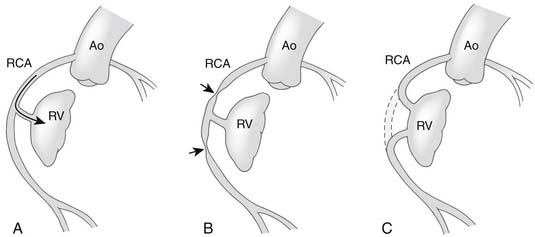
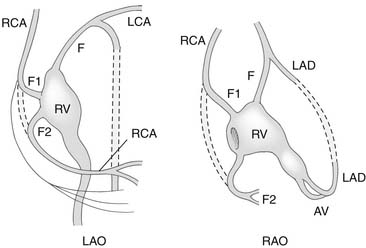
An important anatomic feature of PA/IVS is the presence in some patients of connections between the right ventricle and the coronary circulation (Figs. 113-2 and 113-3). Sinusoids or “intermuscular spaces” in the right ventricular myocardium occur in about 50% of patients.4 In 90% of these patients, the sinusoids communicate with the coronary arteries. The smaller the tricuspid valve (and thus the right ventricular cavity), the more likely it is that right ventricle–coronary arterial fistulas are present. In 15% of patients with these fistulas, significant proximal coronary artery stenoses exist, making myocardial blood flow dependent on blood from the right ventricle (see Table 118-2).10 Knowledge of the presence of a right ventricle–dependent coronary circulation (RVDCC) is important in deciding on surgical therapy, because in these cases, decompression of the right ventricle may cause myocardial ischemia or infarction.14
PATHOPHYSIOLOGY
In PA/IVS, desaturated systemic venous blood is obliged to cross the interatrial septum to mix with saturated pulmonary venous blood in the left atrium (see Fig. 118-1). The resultant admixture is ejected into the systemic arterial circulation, and the systemic arterial saturation depends on adequate pulmonary blood flow. Closure of the ductus arteriosus soon after birth markedly reduces pulmonary blood flow, and progressive hypoxemia and tissue acidosis leads to death. Expeditious administration of prostaglandin E1 can temporarily reverse ductal closure until a surgical procedure to increase pulmonary blood flow can be performed.
INITIAL CLINICAL PRESENTATION AND MANAGEMENT
Infants with PA/IVS are typically full-term, well-developed infants without other anomalies. Delivery is usually uncomplicated, but cyanosis develops on the first day of life and rapidly progresses to respiratory distress and metabolic acidosis. A murmur is unusual unless significant tricuspid regurgitation exists. There is no splitting of the second heart sound. Chest radiography demonstrates clear lung fields with decreased vascular markings. The electrocardiogram is often normal, although the typical neonatal pattern of right ventricular hypertrophy may be absent. Definitive diagnosis is made using two-dimensional echocardiography, which reveals the right ventricular outflow obstruction and the size of the right ventricle and tricuspid valve and, combined with color flow Doppler techniques, can identify right ventricle–coronary artery fistulas.15,16
As soon as the diagnosis of PA/IVS is suspected, an infusion of prostaglandin E1 is begun. Elective intubation and controlled ventilation may be advisable, especially if the infant is to be transported to a tertiary treatment center, because apnea is a common complication of prostaglandin E1 infusion. Cardiac catheterization and cineangiography are recommended for the majority of these patients, especially for those with moderate or severe right ventricular hypoplasia. Right ventriculography, aortography, and, when indicated, selective coronary angiography are performed to determine the presence and extent of right ventricle–to–coronary artery fistulas and the presence of coronary artery obstructions (see Figs. 113-2 and 113-3).14,17 At catheterization, an adequate atrial communication must be ensured. If echocardiography and catheterization indicate that right ventricle–to–pulmonary artery decompression cannot be performed and a flow gradient exists between the right and left atrium, a balloon atrial septostomy is performed at this catheterization.
SURGICAL MANAGEMENT
The ideal long-term outcome for all infants with PA/IVS would be to achieve a two-ventricle circulation with the right ventricle providing all blood flow to the lungs at a low filling pressure without residual right-to-left shunt. The anatomic heterogeneity of this group of patients, however, prevents the achievement of this goal in all patients. In fact, this ideal is achieved in only one third of patients surviving infancy (see Fig. 118-11).10 A more realistic outcome for the remaining patients is the elimination of cyanosis by separating the systemic and pulmonary circulations without limiting cardiac output or inducing excessively elevated systemic venous pressures. Such an outcome can be achieved with a one-ventricle repair (the Fontan operation) for hearts whose right ventricle cannot contribute to pulmonary blood flow, or with a one-and-one-half-ventricle repair for hearts whose right ventricle can provide a portion of pulmonary blood flow. Careful assessment of right ventricular size and coronary artery anatomy are crucial to selecting the appropriate strategy for each patient.
In neonates, it is useful to classify right ventricular hypoplasia into mild, moderate, and severe degrees (Table 118-1). Echocardiographic measurement of the diameter of the tricuspid valve with conversion to a Z-value provides a quantitative measurement to facilitate classification (Fig. 118-4).18 A website (www.parameterz.com) is available for rapid and easy determination of Z-scores. Patients with mild right ventricular hypoplasia have tricuspid valve Z-values of −2 or greater. Tricuspid Z-values between −4 and −2 indicate moderate right ventricular hypoplasia, and Z-values of −4 or less are seen in patients with severe right ventricular hypoplasia. Tripartite right ventricles with a well-developed right ventricular outflow tract fall into the mild group, whereas unipartite right ventricles without a definable infundibular or trabecular portion are classified as severe.19 Patients with mild right ventricular hypoplasia have definite potential for conversion to a two-ventricle system at the time of definitive repair, whereas those with severe right ventricular hypoplasia can achieve only separation of systemic and pulmonary circulations with a one-ventricle repair (a Fontan operation). Patients with moderate right ventricular hypoplasia are also potential candidates for two-ventricle or one-and-one-half-ventricle repair, provided they do not have a right ventricle–dependent coronary circulation.
Table 118–1 Effect of Degree of Right Ventricular Hypoplasia on Surgical Management of Pulmonary Atresia with Intact Ventricular Septum
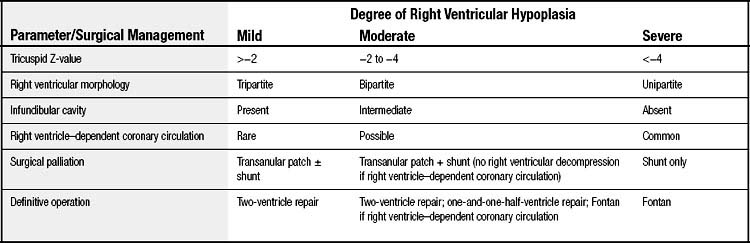
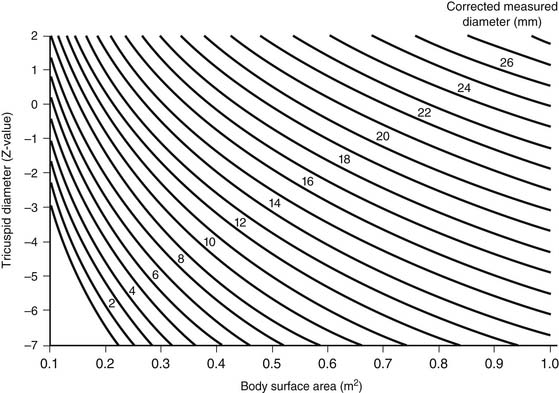
Figure 118–4 Nomogram for converting echocardiographically measured tricuspid valve diameters to the Z-value. Knowledge of the tricuspid Z-value may help select initial surgical management and predict long-term outcome. The Z-value can also be found at www.parameterz.com.
(Modified from Hanley FL, Sade RM, Blackstone EH, et al. J Thorac Cardiovasc Surg 1993;105:406-27.)
Initial Palliation
The surgical management of PA/IVS is typically undertaken in two stages, the first for palliation and the second for definitive repair. To ensure early survival, pulmonary blood flow must be maintained after ductal closure. Although the creation of a systemic-to-pulmonary shunt can ensure continued pulmonary blood flow, the right ventricle must be assessed for its potential recruitment into the circulation. When possible, decompressing the right ventricle into the pulmonary artery permits right ventricular growth such that a two-ventricle repair may become feasible. Failure to decompress the right ventricle at initial palliation essentially eliminates the possibility of ever achieving a definitive two-ventricle repair.20
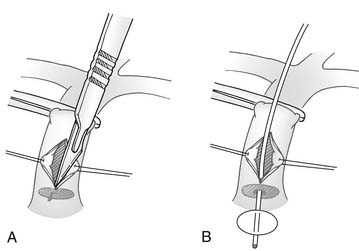
Thus, neonates with mild to moderate right ventricular hypoplasia are best served by relieving the right ventricle–to–pulmonary artery obstruction and creating a systemic-to-pulmonary shunt. This may be accomplished with or without cardiopulmonary bypass. Without bypass, a pulmonary valvotomy can be performed blindly with a transventricular dilator,21 or under direct vision through the main pulmonary artery (Fig. 118-5).22 The use of cardiopulmonary bypass allows more controlled access into the right ventricular outflow tract so that obstructing infundibular muscle can be excised under direct vision and a transanular outflow patch can be placed (Fig. 118-6). By maximizing unobstructed forward flow, transanular patching provides the greatest possibility for right ventricular growth.4,23 Because early postoperative right ventricular failure may cause increased right-to-left shunting across the patent foramen ovale, and antegrade flow from the right ventricle may be limited, a systemic-to-pulmonary artery shunt (a 3- or 3.5-mm polytetrafluoroethylene [PTFE] tube graft) also should be placed to prevent life-threatening hypoxia. Initial results from the Congenital Heart Surgeons Society’s study suggest that concomitant insertion of a transanular patch and placement of a systemic-to-pulmonary artery shunt is the optimal initial treatment for neonates with tricuspid valve Z-values between −1.5 and −44 (Fig. 118-7). In that study, when a valvotomy or transanular patch was performed without a concomitant systemic-to-pulmonary artery shunt, approximately 50% of the patients required shunt placement within the first 4 weeks after the initial procedure. In addition, approximately 40% of patients treated initially with pulmonary valvotomy required a transanular patch at a subsequent operation.4 On the other hand, any form of right ventricular decompression is contraindicated if a right ventricle–dependent coronary circulation exists.
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


