Fig. 18.1
Chest CT scan of a patient with autoimmune PAP. Reticulations are superimposed on ground-glass opacities forming a “crazy paving” pattern with a geographic distribution: juxtaposition of healthy and sick zones
Chest CT Scan
The pattern observed is highly suggestive of the disease, although not pathognomonic. Main abnormalities are ground-glass opacities, septal reticulations and parenchymal consolidation (Fig. 18.1). Reticulations are frequently superimposed on ground-glass opacities, thus forming a “crazy paving” pattern characteristic of PAP. Opacities have a typically geographic distribution, with juxtaposition of healthy and sick zones. The zonal distribution is usually not specific; however, a lower zone predominance might be present in 22 % of cases [16]. Large focal parenchymal consolidations, pulmonary nodules, and mediastinal adenomegaly are usually absent and should lead to the search for an opportunistic infection [16].
Correlation of histology and radiology findings reveals that ground-glass opacities correspond to a lipoproteinaceous alveolar accumulation. Correlations with reticulation are less unequivocal and could correspond to interstitial disease (lipoproteinaceous interstitial accumulation, inflammation or oedema) or lipoproteinaceous alveolar accumulation on the edges of the lobules [17].
A crazy-paving pattern is not specific for PAP and might be associated with lesional or cardiogenic pulmonary oedema, alveolar haemorrhage, pulmonary infection (mycoplasma, pneumocystis), exogenous lipoid pneumonia or bronchioloalveolar carcinoma [17].
The extent of opacities seen on CT scan is associated with impaired pulmonary function on testing [18].
Bronchoalveolar Lavage Fluid
BALF staining is required for the diagnosis of PAP [19]. When performed in a diseased lung area, BALF typically has a milky appearance (Fig. 18.2) but might appear abnormal or normal if performed in a healthy zone, with a weak amount of lipoproteinaceous material.

Fig. 18.2
Bronchoalveolar lavage fluid (BALF) with a milky appearance in PAP (b) as compared to normal saline (a)
Cytologic examination and periodic acid Schiff (PAS) staining are mandatory for diagnosis. In one series, BAL cellularity was increased (330,000 cells/ml) with increased proportion of lymphocytes (mean 57 %) [20]. Careful examination reveals large, foamy macrophages containing eosinophilic granules, with extracellular globular hyaline material found homogeneously positive on PAS and negative on Alcian blue staining (Fig. 18.3).
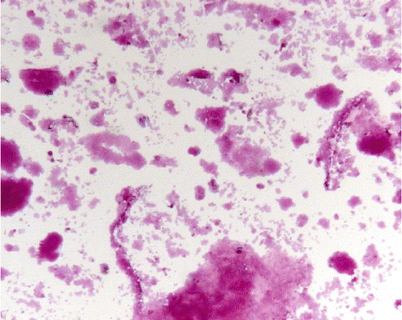
Fig. 18.3
Periodic acid Schiff (PAS) staining of BALF with PAP. Extracellular globular hyaline material homogeneously PAS+, with large, foamy macrophages containing eosinophilic granules
Ultrastructural analysis of BAL fluid is not necessary. It reveals numerous lamellar bodies with a structural resemblance to myelin [7].
Anti-GM-CSF Antibodies
The dosage of anti-GM-CSF antibodies makes the diagnosis of autoimmune PAP. Anti-GM-CSF antibodies may be detected at low concentration in healthy subjects and could participate to the regulation of myeloid cells [21]. Anti-GM-CSF antibodies of IgG, IgA or IgM isotypes have been detected in sera from patients with acute leukemia, their concentration being associated with disease activity [22]. A concentration greater than 19 μg/ml is specific to autoimmune PAP, and a concentration lower than 10 μg/ml has a good negative predictive value [23]. Conflicting data have been published regarding the concentration, the change of auto-antibody levels and evolution of the disease.
Other Biological Exams
Results of routine biological tests are usually normal. Serum lactate dehydrogenase (LDH) may be increased between two and three times the normal range in half of the cases. Increased LDH may be correlated to the severity of the disease. The serum levels of carcinoembryonic antigen and KL-6 (Krebs von den lungen-6) could be higher than those for other diffuse interstitial pneumonia and could be associated with disease severity [12]. Initial level of KL-6 correlates with disease progression and to the need of further specific therapy with a positive predicted value of 91 %.
The serum levels of the surfactant proteins SP-A, −B and -D are increased and could be associated with disease severity [12]. However, levels of SP-A and -B do not change with therapy [24]. In contrast, the level of SP-D in non-human primates was helpful in screening PAP development after injection of anti-GM-CSF antibodies. Determining the level of SP-D could help monitor human disease [10].
Although PAP is autoimmune in 90 % of cases, autoimmune PAP is rarely (<2 %) associated with another autoimmune disease [7].
Pulmonary Function Tests and Exercise Capacity
Pulmonary function and exercise capacity tests are important for therapeutic decisions. Spirometry frequently shows a restrictive pattern but may give normal results in 10–30 % of cases. The most constant and significant modifications are hypoxemia and reduced diffusing capacity of the lung for carbon monoxide (DLCO) with increased alveolar–arterial gradient [11, 14]. Therapy may be introduced in patients showing desaturation on the 6-min walking test.
Open-Lung Biopsy
Special attention to BALF, associated with clinical and CT-scan typical presentation, is often sufficient for diagnosis, and open-lung biopsy is not necessary for diagnosis [11]. Transbronchial biopsy may be helpful. In a Japanese cohort of 203 autoimmune PAP, open-lung biopsy was performed for diagnosis in 8 % of the cases and transbronchial biopsy in 42 % of the cases [12].
Prognosis and Evolution
From spontaneous remission to death, disease evolution is unpredictable. Most recently reported cohorts described a spontaneous remission between 5 and 7 %, lower than the 30 % historical reported. Active smoking is a demonstrated factor of aggravation. Since the wide use of therapeutic lavage, 5-year survival with autoimmune PAP is almost 95 % [12].
Evolution to pulmonary fibrosis is possible after a diagnosis of autoimmune PAP. The risk of fibrosis may be higher in patients who describe toxic inhalation. Non-haematological cancers have been reported, but these associations may be a coincidence [12].
Therapy
The standard of care is symptomatic whole-lung lavage. Numerous therapies targeting an enhancement of the surfactant clearance have been investigated, either targeting alveolar macrophages with exogenous GM-CSF or aiming at reducing levels of anti-GM-CSF antibodies with plasmapheresis or rituximab.
Whole-Lung Lavage
Numerous techniques have been reported.
The first method reported, in 1961, is no longer used. Saline was blindly injected through a percutaneous transtracheal endobronchial catheter. The fluid was re-aspirated through the catheter and evacuated by violent coughing [25]. Classical therapeutic BAL is performed under general anaesthesia in an operating room or an intensive care unit. The patient is intubated with a double-lumen endotracheal tube. The patient under curarisation is placed in the dorsal or lateral decubitus position, with the lung being lavaged in the uppermost position. The non-lavaged lung is mechanically ventilated. One liter of warmed (37 °C) saline is injected in the lung. Fluid is then collected by gravity after opening the outflow tube. Manual or mechanical chest percussion might be performed to improve drainage. The process is repeated until the fluid becomes less opaque; 15 L of saline are generally necessary. The patient is extubated a few hours later depending on the clinical evolution. The controlateral lung may be lavaged 24–48 h later [14, 26].
The most frequent complications are low oxygen saturation, convulsions, pneumothorax, pleural effusion, and fever, which may reveal infection. Retrospective data suggest that whole-lung lavage could improve survival [7]. In 85 % of cases, symptomatic, radiographic and functional improvement is obtained after whole-lung lavage: a mean improvement in FEV1 of 0.26 L, in vital capacity of 0.5 L, in DLCO of 4.4 mL/mmHg/min, and in PaO2 of 20 mmHg, and a mean reduction of alveolar-arterial gradient of 30 mmHg.
According to the centers, between 54 and 90 % of the patients received whole lung lavage. Between 30 and 50 % will need to repeat the whole-lung lavage, on average only one [7]. Almost 10 % of the patients require repeated lavage.
GM-CSF Supplemental Therapy
GM-CSF (Sargramostim®) may be inhaled or subcutaneously administered [27, 28]. Posologies vary from 250 μg to 18 μg/kg/day, fixed or increased dosage, compassionate use or clinical trial.
A meta-analyse had been recently performed with all published data. GM-CSF could be more effective when inhaled, with a response rate at 76.5 % (95 % CI [34.5–95.3]), and a relapse rate of 12.5 % [1.4–64.8]. The response rate of subcutaneous GM-CSF is 48.4 % [33.8–63.3] with a relapse rate of 43.8 [11.8–82.1] after GM-CSF withdrawal. Side effects were considered minor and included injection-site edema, erythema, malaise and shortness of breath.
In the GM-CSF responder group, improvement is slower than after whole-lung lavage. Improvement of PaO2 could be of the same magnitude with both therapies, with an increase of 12–19 mmHg with whole-lung lavage [29, 30] and a mean increase of 23 mmHg with GM-CSF therapy in GM-CSF-responder patients, for 9.7 mmHg for all GM-SCF patients [27].
No clinical or biological marker exists to predict response to GM-CSF and to select patients that could benefit from GM-CSF therapy. In one study, factors associated with response to subcutaneous GM-CSF therapy were hypereosinophilia under therapy, longer delay since diagnosis, higher vital capacity, normal serum LDH concentration and increased serum SP-B concentration [27]. Indeed, the initial concentration of anti-GM-CSF antibody and the evolution of concentration under therapy are not associated with response [7, 27, 31]. Low vital capacity at baseline may be associated with relapse and the need for an additional treatment [32].
GM-CSF therapy is now considered an alternative to whole-lung lavage. However, Sargramostim® was withdrawn from several national markets and may require specific authorisation from national health authorities according to each country.
Rituximab and Plasmapheresis
Immunosuppressive therapies, particularly corticosteroids, are not effective in PAP [7] and could increase the risk of pulmonary infection. Theoretically, plasmapheresis should be effective to decrease the concentration of anti-GM-CSF antibodies and improve the disease as in Goodpasture disease [33]. Only two cases have been reported in the literature: a decrease of anti-GM-CSF antibodies was measured in both cases, and one patient showed a striking improvement and the other a mild improvement [34, 35].
Rituximab, a monoclonal antibody directed against the CD20 antigen of B lymphocytes, could ameliorate PAP by decreasing anti-GM-CSF antibody concentration. Two cases have been reported in the literature of rituximab efficacy [36, 37]. A prospective monocentric open-label study evidenced an improvement in seven of the nine treated patients with rituximab associated with a decrease of serum anti-GM-CSF antibody concentration [38]. Most patients received 1,000 mg of Rituximab, days 1 and 15. Mean increase of PaO2 was 12 mmHg 3 months after therapy. Rituximab was well tolerated in all patients.
Although the exact place of these treatments is not well defined yet, rituximab therapy could be an alternative for whole-lung lavage resistant disease.
Secondary PAP
Secondary PAP includes PAP secondary to immune deficiency, cancer and particularly haematological diseases, and secondary to toxic inhalation.
Immune Deficiency
PAP has been rarely associated with immune deficiency, including severe combined immunodeficiency, agammaglobulinemia [39], or organ transplantation (Fig. 18.4) [7]. A few cases of connective tissue diseases have been reported: dermatomyositis [40], rheumatoid arthritis [41], and Behcet’s disease [42, 43]. The, recently described, MonoMAC syndrome include monocytopenia, mycobacterial diseases and frequent PAP [44]. MonoMAC syndrome is associated with GATA2 mutations and will be described further.
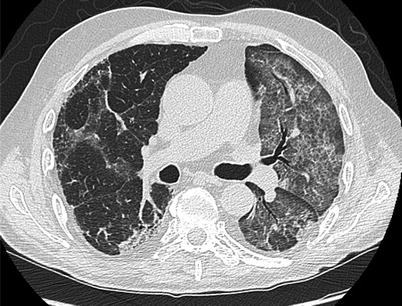
Fig. 18.4
Chest CT of a patient that developed PAP after lung transplantation CT shows ground-glass opacities and reticulations. PAP resolved with modification of immunosuppressive therapy
Cancer
The association of PAP and haematological disorders is well established, mostly myelodysplastic syndromes (8–74 % of the cases) and acute myeloid leukaemia (5–21 %) [43]. Some cytogenetic abnormality such as trisomy 8 could be more frequently associated with PAP. PAP could explain up to 10 % of pulmonary manifestations during these diseases [45]. Less frequently, PAP has been associated with acute lymphoid leukaemia [46], lymphoma [47], and myeloma [48]. In haematological diseases, alveolar macrophages could be numerically or functionally unable to clear the surfactant.
PAP is generally diagnosed during the evolution of haematological disease and may occur in the absence of detectable tumour after bone marrow transplantation [49]. The diagnosis of PAP and haematological disease may occur simultaneously; in that case, the diagnosis of the haematological disease is generally easy [43, 50].
The classical presentation is respiratory insufficiency with fever in 24 % of the cases, particularly in patients with prolonged neutropenia secondary to chemotherapy [45]. However 20 % of the patients may be asymptomatic at the diagnosis.
Chest X-ray shows diffuse interstitial opacities [45]. Chest CT scan may show the typical crazy paving pattern. CT scan may also be less suggestive by showing ground-glass opacities mainly in the lower zone without subpleural sparing or consolidations (Fig. 18.5) [47, 51]. Fiberoptic bronchoscopy is diagnostic. Interestingly, BALF analysis may reveal PAP and an associated opportunistic infection. Pulmonary infection with Nocardia, Pneumocystis, Acinetobacter, Aspergillus, Cladosporium, or Mycobacerium tuberculosis or non-tuberculosis Mycobacteria have been associated with PAP [47, 52]. Positive PAS staining of intra- and extracellular material is sufficient for the diagnosis of PAP and may negate a lung biopsy or autopsy [45, 53]. In a retrospective study of 40 cases, the diagnosis was made by BALF analysis in 21 cases, by transbronchial biopsies in 9 cases and by surgery in 10 cases.
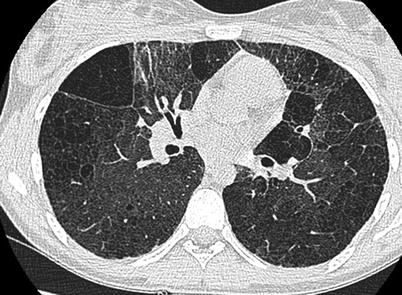
Fig. 18.5
Chest CT scan of a patient with interstitial lung disease associated with ABCA3 mutation PAP. CT scan shows diffuse ground-glass opacities and lung cysts
The prognosis is poor and the median time of survival of less than 20 months. In a retrospective study of 35 patients and in a review of 44 patients, the causes of deaths were: haematological disease (33 %), infection (29 %), and respiratory insufficiency (25 %).
Less than 20 % of patients who benefited from whole-lung lavage showed improvement [54]. Haematological therapy alone, chemotherapy and/or bone-marrow transplantation, can cure PAP, particularly in cases of acute leukaemia [55, 56].
PAP may be associated with non-haematological cancers, most frequently lung cancers (squamous cell cancer (N = 2), adenocarcinoma (N = 2)) and more rarely, mesothelioma, melanoma with lung metastasis and glioblastoma (N = 1 each) [57]. For at least one of these cases, anti-GM-CSF antibodies were detected. This association could be a coincidence in view of the frequency of lung cancer.
Inhalation of Toxic Particles
Numerous case reports or short series describe PAP after inhalation of mineral particles (silica, talc, cement, kaolin), metal particles (aluminium, titanium, indium), or more rarely organic particles (fibres of cellulose) [7, 11, 58]. PAP may occur after massive inhalation of silica and is then called acute silicoproteinosis [7]. Clinical presentation is close to that of autoimmune PAP.
Numerous animal models demonstrated the development of PAP after inhalation of nickel, silica, titanium, quartz, fibreglass, indium or aluminium powder [11, 59, 60]. In the French series reported by Briens et al., in 2002, 39 % of patients reported notable professional exposure to particles [14]. Furthermore, in the Japanese cohort of 248 patients with autoimmune PAP, 23 % were considered exposed to toxic inhalation [12]. However anti-GM-CSF antibodies were detected in a patient with PAP secondary to indium inhalation [58]. This suggests that some of the secondary PAP may be associated with anti-GM-CSF antibodies and that toxic inhalation could be a trigger for an autoimmune disease [61].
Clinical presentation is close to that of the autoimmune PAP, but evolution may be more severe. In a Japanese series of 88 patients, 15 were dead. Causes of death were: respiratory insufficiency including pulmonary fibrosis (n = 8), cancers (n = 3), infections (n = 2); Median survival was 17.2 years. Persistent toxic inhalation was associated with a worse prognosis [62].
Genetic PAP
Genetic PAP could be divided in two groups:
Mutations that involved the production of surfactant SFTPB, SFTPC, ATP-binding cassette 3 (ABCA3) and NK2 homeobox 1(NKX2–1).
Mutations that involved the macrophage and the degradation of surfactant, GM-CSF receptor, α or β subunit, mutations of GATA2 as well as lysurinic protein intolerance.
Moreover, a large kindred of 32 patients with PAP originated from La Reunion island has been recently described [63]. The diseases occurred in the first 6 months of life with a 59 %, 5 years mortality. Associated liver diseases were frequent. To date, the very probable mutated gene has not been described.
Mutations of SFTPB, SFTPC, ABCA3 and NKX2–1
The term of PAP has been widely used in patients with surfactant mutation associated disorders [64]. Indeed, both entities share similar clinical characteristics which probably explain the confusion [65]. The radiological and the histological presentation are nonetheless different. In both children and adult, diffuse ground-glass opacities and lung cysts are the common radiological features observed in surfactant protein disorders whereas septal reticulations, and parenchymal consolidation usually described in autoimmune PAP are less frequent (Fig. 18.6) [66, 67]. The histopathologic findings in infants with mutations in the SFTPB, SFTPC, ABCA3 or NKX2.1 genes are remarkably similar, demonstrating varying degrees of interstitial thickening, a remodelling of the alveolar epithelium with type II cell hyperplasia, as well as alveolar accumulation of eosinophilic, lipoproteinaceous, granular material [66–69].
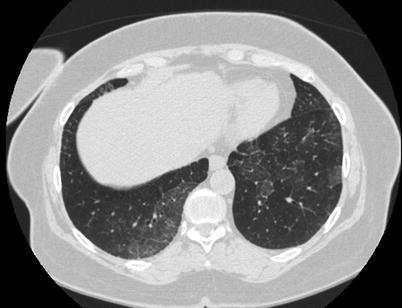
Fig. 18.6
Chest CT scan of a patient with PAP secondary to myelodysplastic syndrome. CT shows ground-glass opacities and reticulations mainly in the lower zone with patchy, slightly peripheral distribution
Genetic Defect in GM-CSF Receptor
GM-CSF receptor is composed of the binding α chain (CD116), coded by CSF2RA, and the common β chain (CD131), coded by CSF2RB, which is also shared by IL-3 and IL-5 receptors.
Mutations of CSF2RA have been described only in children, and a series of eight patients from 1.5 to 9 years was recently reported [72]. Mutations of CSF2RA could correspond to 6 % of all PAP [72]. CSF2RA is located on X and Y chromosomes. The transmission is autosomal recessive. Very recently the mutation was described in a 3 year patient with PAP after a complex inactivation of the naïve gene in the other X chromosome [73]. Some mutations have varying and incomplete penetrance, and disease was found in three asymptomatic children between 5 and 8 years after the diagnosis of a familial index case [74].
Except for a lower age at disease onset, patients with mutation of CSF2RA present PAP close to autoimmune PAP. Indeed, unlike mutations of surfactant proteins, interstitial cell infiltration is absent. However, alveolar and serum concentration of GM-CSF is increased, and anti-GM-CSF antibodies are absent.
Mutation of CSF2RB was suggested in three patients presenting neonatal PAP and confirmed in a 36 years woman and in 9 years girl [75–77]. Both showed disease close to autoimmune PAP without detectable anti-GM-CSF antibodies but with high concentration of GM-CSF [77].
Lysinuric Protein Intolerance
Lysinuric protein intolerance is an autosomal-recessive disease caused by mutation of SLC7A7 contributing to defective transport of cationic amino acid at the membrane of epithelial cells in the intestine and kidney. In Japan, the estimated prevalence is 1/57,000 births. The clinical presentation is characterized by failure to thrive and gastrointestinal symptoms. The most frequent chronic manifestations are related to renal and pancreatic insufficiency.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


