This chapter will consider those progressive pediatric encephalopathies that present with seizures where electroencephalography (EEG) plays a large role in the diagnostic evaluation. The EEG can be useful in several ways. A careful examination of the background can help to confirm the clinical suspicion of an encephalopathy, even in mildly affected children. Furthermore, if serial studies are performed, deterioration of the background may indicate a progressive encephalopathy. Distinguishing static from progressive illness is a very useful clinical distinction, but EEG provides more information than just confirmation of a progressive encephalopathy. There are certain EEG patterns that are highly suggestive and should prompt an evaluation for an inborn error of metabolism (IEM). In rare and selected circumstances, the EEG findings may suggest a specific biochemical cause, or a discrete list of conditions. In addition to these diagnostic considerations, the EEG may be one of the most sensitive measures to gauge improvement in those situations where effective treatment is possible (e.g., pyridoxine-dependent epilepsy).
Limited Repertoire of the Immature EEG
There is a very important caveat that must be stated before any further discussion: in the majority of circumstances, the EEG features are not specific for any particular disorder. Considering that there are over 200 IEMs associated with seizures, the range of EEG abnormalities seen in clinical practice is remarkably constrained. This may relate to the limited repertoire of the EEG expression in the very young or to shared pathophysiological mechanisms in the advanced stage of many diverse conditions. While one might expect to see a much greater repertoire of abnormalities, experience teaches us otherwise; even the most skilled pediatric electroencephalographers cannot suggest a specific diagnosis in most patients with IEMs simply by examining the EEG.
The EEG Hallmark of a Pediatric Progressive Encephalopathy
There is, instead, a remarkably similar pattern of EEG abnormalities in most cases: the background is slowed; there is lack of the expected organization for age; multifocal spikes are present; and sometimes normal physiological hallmarks, like sleep spindles, are lost. This pattern is etiologically nonspecific and is frequently found in children with epileptogenic encephalopathies (see pattern 4a in Chapter 10), of which only a subset will be caused by IEMs.
The most severe disorders will produce even more dramatic changes with some degree of discontinuity in the tracing. In certain cases, this may only be apparent in the sleep tracing, but in the most severe, it will be present in awake and asleep. In these conditions, the epilepsy itself may contribute to the encephalopathy and they are referred to as “epileptic encephalopathies” (EEG pattern 4b in Chapter 10). There is a spectrum of these disorders where the precise clinical and electrographic manifestations are age-dependent. In neonates with severe conditions, a burst-suppression pattern is seen, whereas in infancy, hypsarhythmia predominates. It is not until childhood that well-formed generalized spike-wave discharges (SWDs) are seen.
A Red Flag for IEMs: Spectrum of Epileptic Encephalopathies
The most important EEG patterns indicative of an IEM are seen in the spectrum of severe epileptic encephalopathies. These extend from Ohtahara syndrome (early infantile epileptic encephalopathy or EIEE), to West syndrome (WS), onto late infantile epileptic encephalopathy (LIEE), to Lennox-Gastaut syndrome (LGS). This concept is explained in detail in Chapter 10. In these conditions, the electro-clinical expression is largely dependent on the age of the child, but there are several common EEG features. In nearly all cases, the backgrounds will be slowed and there will be multifocal spikes with variable morphology (pleomorphic multifocal spikes). In addition, the background will show some degree of discontinuity. In the most severe cases, it is a burst-suppression pattern, particularly with those patients who present in the neonatal and early infantile period. Those who present slightly later, approximately 6 months of age and greater, will often have hypsarhythmia. It is rare to see a child presenting with LIEE and LGS as the manifestation of their IEM—they usually have symptoms before then, or conversely, milder conditions that present with less dramatic EEG features. Children presenting later in childhood or adolescence often have multifocal or generalized spikes superimposed on slowed but continuous backgrounds.
Given all this, it is helpful to organize progressive pediatric syndromes by age of presentation, highlighting prototypic EEG presentations in each age group. Wherever possible, we have underscored specific EEG features that are particularly declarative. For a comprehensive listing or further details, the interested reader is referred to resources such as Online Mendelian Inheritance in Man (OMIM—www.ncbi.nlm.nih.gov/omim).
NEONATAL
There are four well-characterized epilepsy syndromes presenting in the newborn; two have largely normal EEG backgrounds and are not associated with IEMs: benign neonatal convulsions and benign familial neonatal convulsions. Two others have markedly disorganized backgrounds with burst suppression: early myoclonic encephalopathy (EME) and Ohtahara syndrome. EME is the prototypic epilepsy syndrome in this epoch of life associated with inborn errors. There are very rare case reports of IEM in Ohtahara syndrome.
EME—A Red Flag for IEM
The neonatal epilepsy syndrome most clearly associated with IEM is EME. This syndrome is characterized by refractory seizures, often myoclonic or epileptic spasms, with a burst-suppression pattern. The burst-suppression pattern often has variable intervals between the bursts and may be state dependent (Fig. 9.1). Published causes of EME include nonketotic hyperglycinemia, methylmalonic academia, propionic academia, D-glyceric academia, sulfite oxidase deficiency/molybdenum cofactor deficiency, Leigh syndrome, carbamyl phosphate synthetase deficiency, pyridoxine dependency, Menkes disease, and peroxisomal disorders. An inspection of this list shows that the majority of these are organic and aminoacidurias, and the most prominent of these is nonketotic hyperglycinemia. One could simplify this list to the following six considerations: organic/amino academies, including nonketotic hyperglycinemia, urea cycle defects, mitochondrial disorders, defects of the peroxisomes, Menkes disease, and pyridoxine dependency (Tables 9.1 and 9.2).
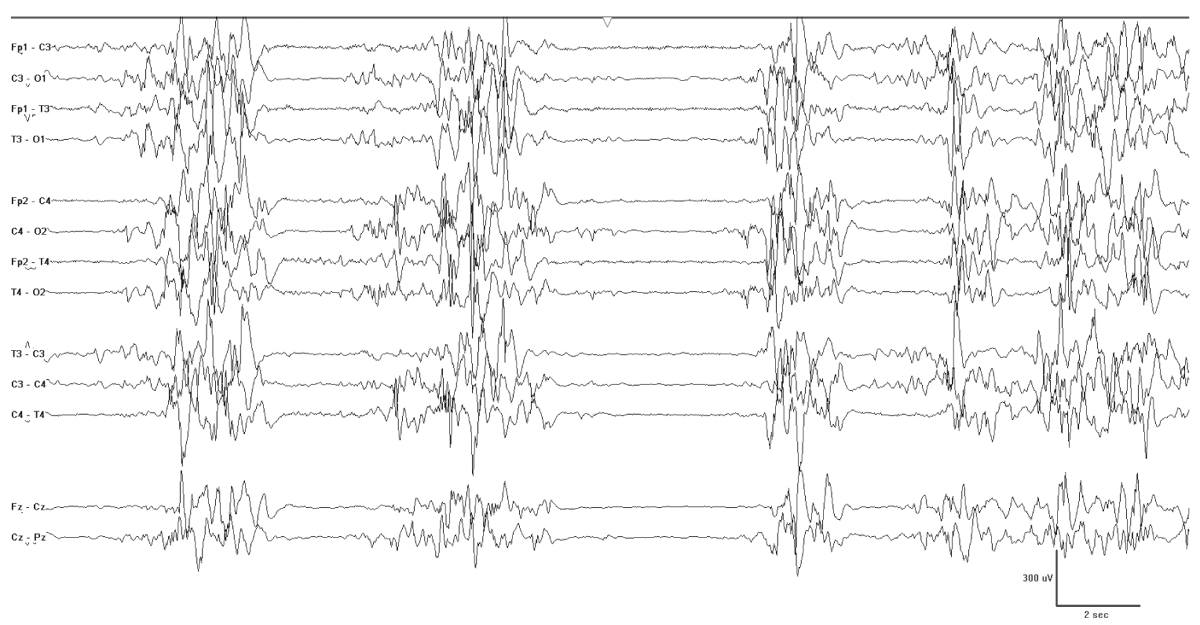
Figure 9.1: EME. This neonate was born at 38 weeks and had erratic myoclonia. Note the burst-suppression pattern characteristic of this condition.
IEMs Causing EME
Organic/amino acidurias |
Urea cycle defects |
Disorders of subcellular organelles Peroxisomes Mitochondria |
Menkes disease |
Pyridoxine dependency |
Important Diagnostic Tests for EME
Complete blood count |
Chemistry profile including liver function tests |
Urine organic acids |
Amino acid profile in CSF, blood, and urine |
Lactate/pyruvate in CSF and blood |
Serum ammonia |
Urine copper, serum cerruloplasmin (check for Menkes) |
Very long chain fatty acids |
Pyridoxine challenge, pyridoxal 5-phosphate challenge, or both |
A Prototypic Cause of EME: Nonketotic Hyperglycinemia or Glycine Encephalopathy
In this autosomal recessive inborn error of amino acid metabolism, also known as glycine encephalopathy, large amounts of glycine accumulate in the body fluids because of a defect in the multienzyme complex for glycine cleavage. This mitochondrial enzyme system has four protein components (P, H, T, and L). The most common mutation causes P protein dysfunction. The pathophysiological processes have not been fully elucidated, but the elevated glycine level is believed to affect the central nervous system through its roles as an inhibitory transmitter in the brainstem and spinal cord and excitatory transmitter acting upon NMDA receptors in the cortex. Glycine may also impair cellular energetics by interfering with citrate synthetase function and mitochondrial electron transport.
From the early descriptions of this disorder in 1965 and 1968, it was apparent that affected neonates usually have lethargy, respiratory difficulties, and seizures in the first 48 hours (1). Hiccoughs may be seen by fourth or fifth day of life. Seizures are often myoclonic or spasms and accompanied by a burst-suppression pattern (2). As patients with these disorders mature, infantile spasms may persist, and the EEG often reveals hypsarhythmia. Cortical malformations or corpus callosum defects may be present. Recently, diffusion-weighted changes have been noted in the acute stages on MRI, which may be suggestive of the disorder (3).
Notably, a transient form of nonketotic hyperglycinemia exists with similar early clinical and biochemical findings. In this condition, however, glycine concentrations normalize between 2 and 8 weeks after birth, and the prognosis is favorable.
The characteristic EEG background is burst suppression. Synchronous diffuse bursts are separated by several seconds of profound background attenuation. The bursts themselves are composed of irregular slow-wave activity with superimposed multifocal sharp transients. The period between the bursts varies from moment to moment. The level of alertness and age of the child can substantially modify the continuity of the tracing (Fig. 9.2).
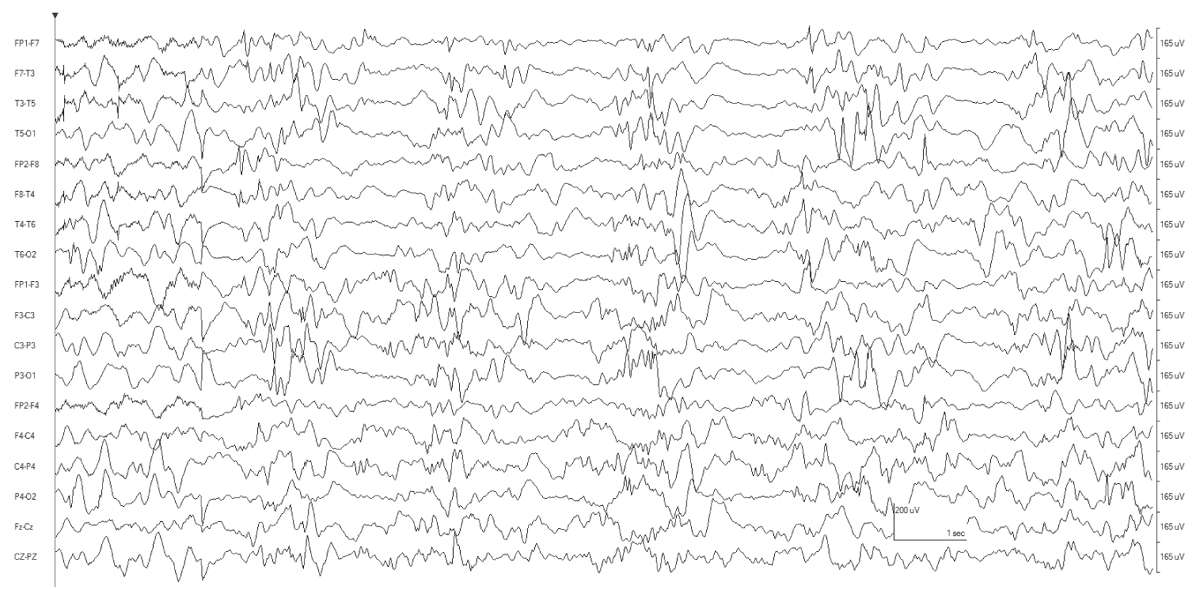
Figure 9.2: This tracing is from a 3-month-old infant with nonketotic hyperglycinemia. There is still some hint of discontinuity to the tracing, but it no longer meets criteria for burst suppression. In addition to myoclonia, the patient also had epileptic spasms (not shown here).
Other Important IEMs Causing Seizures in the Newborn
Some newborns will not have a well-characterized epilepsy syndrome, while others that do not fit into either Ohtahara or EME syndrome can be considered to have an early onset epileptic encephalopathy (EOEE) (4). Still others may show a mixture of focal, myoclonic, and tonic seizures, with backgrounds that are abnormal but do not meet criteria for burst suppression. As a consequence, virtually any neonate with persistent seizures without a clearly identifiable cause should be evaluated for an IEM and the newly discovered channelopathies.
IEMs that present in the newborn include nonketotic hyperglycinemia; isovaleric, methylmalonic and propionic aciduria; maple syrup urine disease; pyridoxine dependency, pyridoxal phosphate dependency; biotinidase deficiency; D-2-hydroxyglutaric aciduria, 3-phophoglycerate dehydrogenase deficiency; Adenylosuccinate Lyase deficiency methylene tetrahydrofolate reductase (MTHFR) deficiency; GABA transaminase deficiency; congenital disorders of glycosylation; developmental delay, epilepsy and neonatal diabetes (DEND); hyperinsulinism, hyperammonemia (HIHA); neonatal ceroid lipofuscinosis with cathepsin deficiency; creatine deficiency; peroxisomal disorders, urea cycle disorders; dihydropyrimidine dehydrogenase deficiency; molybdenum cofactor deficiency; carnitine palmitoyltransferase types I and II; pyruvate carboxylase deficiency; pyruvate dehydrogenase deficiency; and disorders of biotin. In short, every class of metabolic derangement may present in the very young, and most of these have few distinguishing clues (Table 9.3).
Epileptic Encephalopathies in the Newborn
Nonketotic hyperglycinemia |
Isovaleric, methylmalonic, and propionic aciduria |
Maple syrup urine disease |
Pyridoxine dependency, pyridoxal phosphate dependency |
Biotinidase deficiency |
D-2-hydroxyglutaric aciduria |
3-Phosphoglycerate dehydrogenase deficiency |
Adenylosuccinate Lyase deficiency |
MTHFR deficiency |
GABA transaminase deficiency |
CDG |
Developmental DEND |
HIHA |
Neonatal ceroid lipofuscinosis with cathepsin deficiency |
Creatine deficiency |
Peroxisomal disorders |
Urea cycle disorders |
Dihydropyrimidine dehydrogenase deficiency |
Molybdenum cofactor deficiency |
Carnitinepalmitoyl transferase types I and II |
Pyruvate carboxylase deficiency |
Pyruvate dehydrogenase deficiency |
Disorders of biotin |
Pyridoxine Dependency
In this condition, refractory seizures typically develop within the first several days after birth characterized by infantile spasms or a variety of focal, myoclonic, and atonic seizures (5). A recent review highlights the salient features and summarizes the biochemistry (6). The condition is caused by mutations in the antiquitin gene, which codes for an enzyme in lysine metabolism (α-aminoadipic semialdehyde dehydrogenase). As a result a variety of substrates accumulate, including α-aminoadipic semialdehyde (AASA) and L-Δ1-piperideine-6-carboxylate (P6C). The latter undergoes a chemical condensation with pyridoxal 5-phosphate (PLP), thereby lowering PLP free levels. Both AASA and P6C may be neurotoxic in and of themselves, and low PLP levels most likely alter the balance between glutamate and GABA since PLP is a necessary cofactor for glutamate decarboxylase function.
The EEG is characterized by generalized bursts of high-voltage delta with intermixed spike-and-sharp waves and periods of asynchronous attenuation (Fig. 9.3A). Since 1954, it has been known that treatment with high doses (100 to 200 mg intravenously) of pyridoxine can have a dramatic early effect on seizures and the resting EEG (5). There may be immediate cessation of seizures, conversion of the EEG to a burst-suppression pattern, and later normalization of the EEG with subsequent doses (see Fig. 9.3B). Intravenous (IV) treatment or even oral replacement can result in transient apnea; so practitioners should be prepared to support respirations before administering pyridoxine. The onset of this disease may occur after the newborn period, making it a consideration in older infants with refractory seizures of any type.
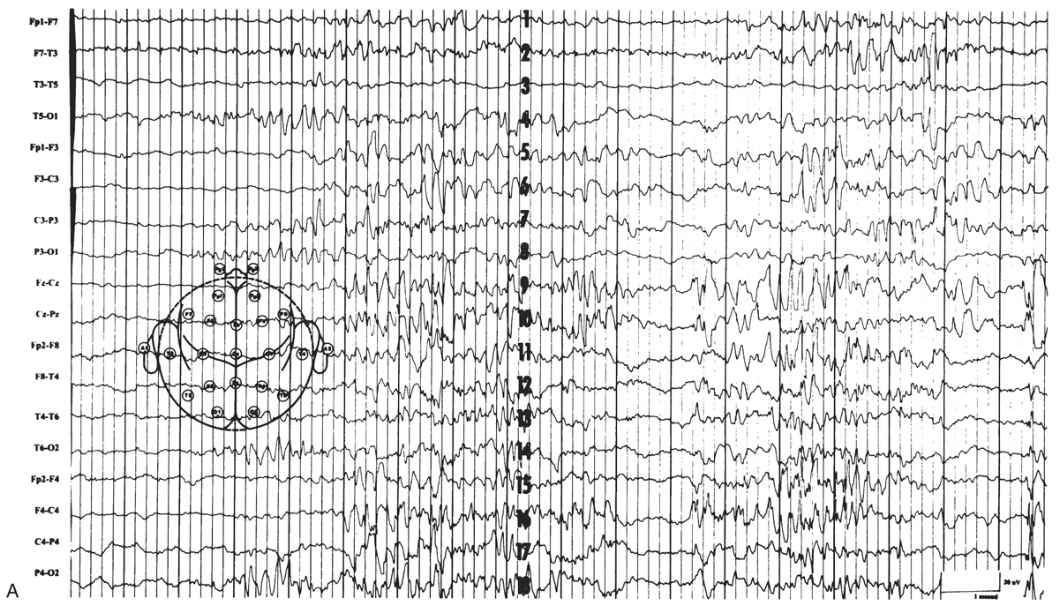
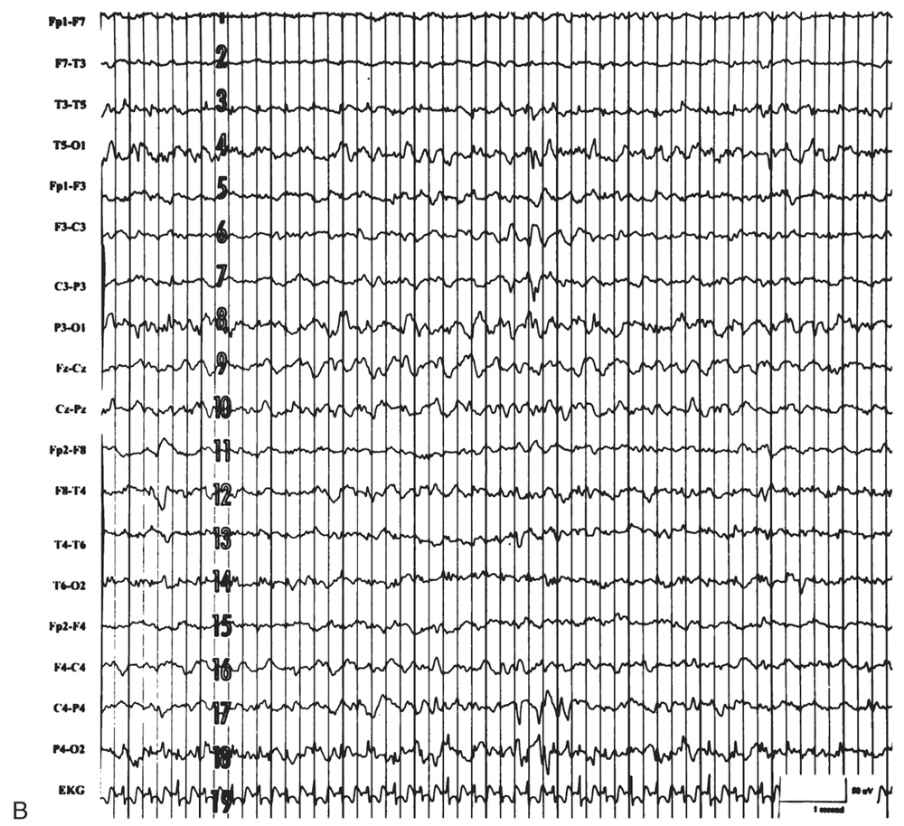
Figure 9.3: Pyridoxine dependency. A: EEG of a 4-week-old child with intractable neonatal seizures. There are bursts of high-voltage delta with intermixed spikes and sharp waves and asynchronous epochs of attenuation. B: EEG of the same child after pyridoxine treatment. The tracing is now continuous, and only occasional epileptiform discharges persist. (Courtesy of C. Stack, MD, and L. Laux, MD, Children’s Memorial Hospital, Chicago, Illinois.)
A related and recently characterized condition is pyridoxal 5′-phosphate-dependent epilepsy (PLP-DE). This is most often due to mutations in the gene encoding pyridoxine 5′-phosphate oxidase (PNPO). Like pyridoxine-dependent epilepsy (PDE), this condition presents with intractable neonatal or infantile seizures, but instead responds only to PLP, the active form of pyridoxine.
Evaluation and Treatment of PDE and PLP-DE
Plasma samples for pipecolic acid as well as urine and plasma samples of AASA should be obtained in at-risk individuals. Some also advocate CSF analysis of the same substances. Treatment regimens vary. The recommended oral dose of either pyridoxine or PLP is 30 mg/kg/day (6,7). Since there is no readily available IV preparation of PLP, some authors administer 100 mg of IV pyridoxine, and if there is no clear EEG response, repeat the dose and then treat with oral PLP while the results of biochemical tests including CSF, serum markers, and urine markers of PDE and PLP-DE return. Suspected cases can be confirmed with genetic mutational analysis of either conditions.
Molybdenum Cofactor Deficiency and Sulfite Oxidase Deficiency
These rare conditions manifest shortly after birth with a progressive encephalopathy, feeding difficulties, hypotonia, and refractory partial, myoclonic, or apparently generalized seizures. Dysmorphic features, lens dislocation, and hepatomegaly are characteristic (8–10). Neuroimaging may show poor differentiation between the gray and white matter, severe cerebral and cerebellar atrophy, and multiple cystic cavities in the white matter (WM). The relatively more common molybdenum cofactor deficiency results from an absence of the hepatic cofactor, which leads to a combined enzyme deficiency of the three molybdenum-dependent enzymes: sulfite oxidase, xanthine dehydrogenase, and aldehyde oxidase. Reduction in sulfite oxidase activity causes an accumulation of sulfite in the brain, which is toxic to its function. This disease may be caused by mutations in the genes MOCS1, MOCS2, or gephyrin (GEPH) (11).
The EEG shows multifocal spikes with or without burst suppression. Burst suppression may evolve in the second day of life and persist despite treatment with anticonvulsant medications (12).
Peroxisomal Disorders
Disorders of the peroxisome have been divided into three categories: disorders of peroxisomal biogenesis (Zellweger syndrome, neonatal adrenoleukodystrophy [NALD], infantile Refsum disease); disorders of a single peroxisomal enzyme (X-linked adrenoleukodystrophy, acyl-coenzyme A [CoA] oxidase deficiency [AOXD]); and disorders with deficiencies of multiple peroxisomal enzymes (rhizomelic chondrodysplasia punctata). This discussion will be limited to Zellweger syndrome, AOXD, and NALD.
Zellweger Syndrome
Zellweger syndrome is the most frequent peroxisomal disorder in early infancy. It is caused by mutations in any of several genes involved in peroxisome biogenesis, resulting in severe brain dysfunction, craniofacial deformities, liver disease, and absence of peroxisomes.
Dysmorphic features may be noted shortly after birth. Within the first week, the affected child develops a severe encephalopathy, hypotonia, and hyporeflexia. Findings on neuroimaging and pathological examination are distinctive, with pachygyria or polymicrogyria localized to the opercular region and cerebellar heterotopias.
Patients with Zellweger syndrome have focal motor seizures of the arms, legs, or face. Their seizures do not usually culminate in generalized seizures. Myoclonic seizures and spasms have also been described. Interictal EEGs of the patients with Zellweger syndrome showed infrequent bilateral independent multifocal spikes, predominantly in the frontal motor cortex and its surrounding regions (13). Less frequently, hypsarhythmia is observed. Some have noted prolonged runs of vertex negative spikes (14,15).
Neonatal Adrenoleukodystrophy
NALD, an autosomal recessive disorder with pathological and biochemical findings resembling those of X-linked adrenoleukodystrophy, bears some similarity to Zellweger syndrome but differs in some respects, including the ability to synthesize plasmalogens in NALD (16). Distinguishing features include absent or minimal facial dysmorphisms, later onset of seizures, absent or minimal cerebral malformations, and longer life span. Described seizures include tonic, clonic, myoclonic, or epileptic spasms (17). No characteristic EEG pattern has been defined, but descriptions have included high-voltage slowing, polymorphic delta activity, multifocal paroxysmal discharges, burst suppression, and hypsarhythmia.
Acyl-Coenzyme A Oxidase Deficiency
AOXD was initially described in two siblings in 1988 (18). Clinical features include hypotonia, pigmentary retinopathy, hearing loss, developmental delay, adrenocortical insufficiency, absence of dysmorphic features, and onset of seizures shortly after birth. Serum levels of very long chain fatty acids are elevated, with normal levels of pipecolic acid. Cortical malformations are generally absent, and the interictal EEG may show continuous, diffuse, high-voltage theta activity.
Urea Cycle Disorders
There are six enzymes in the urea cycle that convert ammonia to urea; five of these may have defects that cause neonatal seizures. The associated diseases and their incidences are as follows: carbamoyl phosphate synthetase deficiency, 1 in 800,000; ornithine transcarbamylase deficiency, 1 in 80,000; argininosuccinate synthetase deficiency, 1 in 250,000; argininosuccinate lyase deficiency, 1 in 70,000; and N-acetylglutamate synthetase deficiency, with only a few cases reported. The clinical disorder resulting from a deficiency in the sixth enzyme, arginase, manifests later in infancy. All of these disorders are autosomal recessive except for ornithine transcarbamylase deficiency, which is X-linked dominant.
The clinical manifestations are similar and result, at least in part, from ammonia elevations. Typically, affected newborns exhibit poor feeding, emesis, hyperventilation, lethargy, or convulsions 1 to 5 days after birth. These signs give way to deepening coma with decorticate and decerebrate posturing and progressive loss of brainstem function. Brain imaging and pathological studies reveal cerebral edema with pronounced astrocytic swelling. Ammonium is taken up by the astrocytes and rapidly converted to glutamine in an energy-dependent process. The edema is associated with glutamine accumulation and energy depletion. In experimental models, blocking glutamine synthesis has prevented cerebral edema associated with hyperammonemia (19).
The EEG shows a low-voltage pattern with diffuse slowing and multifocal epileptiform discharges (Fig. 9.4A), and sometimes sustained monorhythmic theta activity (see Fig. 9.4B) (20). In acute neonatal citrullinemia, a burst-suppression pattern has been described, correlating with the degree of hyperammonemia (21).
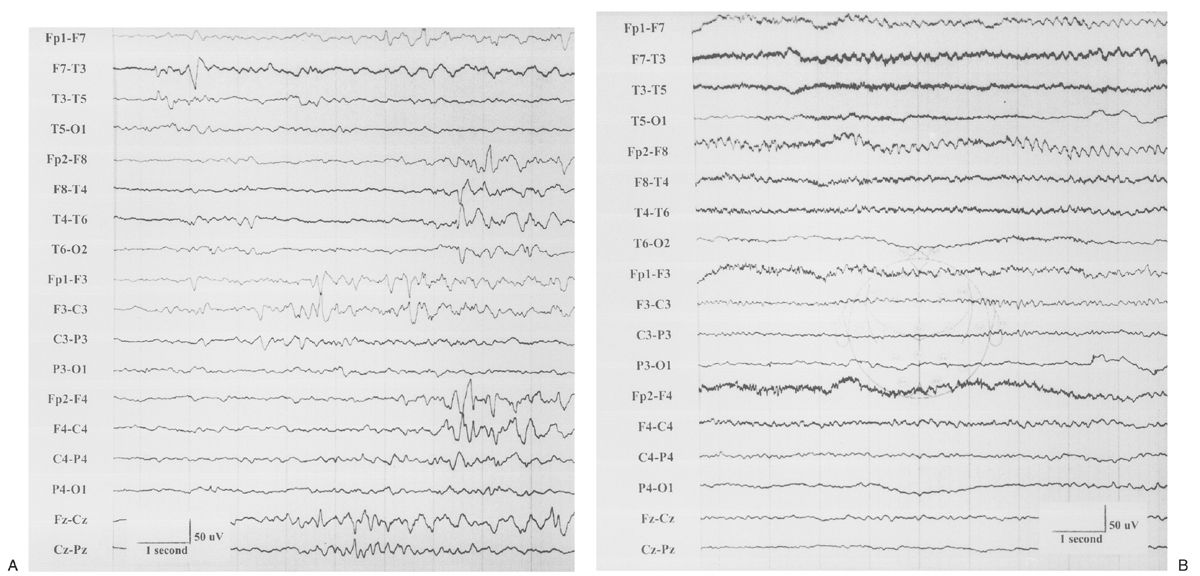
Figure 9.4: EEGs of a 2-week-old child with ornithine transcarbamylase deficiency. A: There is marked attenuation and disorganization of the background with multifocal spikes. B: Sustained monorhythmic theta activity is present in the frontal regions. (Courtesy of C. Stack, MD, and L. Laux, MD, Children’s Memorial Hospital, Chicago, Illinois.)
Maple Syrup Urine Disease
This disease, resulting from a defect in the branched chain α-keto acid dehydrogenase complex, was first reported by Menkes et al. (22) in 1954. The enzyme defect leads to accumulation of the branched-chain amino acids—valine, leucine, isoleucine—and their keto acids in the body tissues and fluids. Pathological studies have revealed diffuse myelin loss and increased total brain lipid content. Cystic degeneration of the WM associated with gliosis is seen. Disordered neuronal migration may occur with heterotopias and disrupted cortical lamination.
Feeding difficulties and lethargy are observed during the first to second weeks after birth. If left untreated, symptoms may progress to stupor, apnea, opisthotonus, myoclonic jerks, and partial and generalized seizures. A characteristic odor can be detected in urine and cerumen, but this may not be detectable until several weeks after birth.
The EEG shows diffuse slowing and a loss of reactivity to auditory stimuli. A distinctive comb-like rhythm has been described, with excessive bursts of a centrally predominant mu-like rhythm, which resolve with dietary therapy (23,24).
Organic Acidurias
The disorders of organic acid metabolism comprise a large number of inborn errors, including isovaleric aciduria and several ketotic hyperglycinemic syndromes (propionic acidemia, methylmalonicacidemia, and α-ketothiolase deficiency).
Symptoms of isovaleric aciduria, which develop during the neonatal period in half of the affected children, include poor feeding, vomiting, dehydration, and a progressive encephalopathy manifested by lethargy, tremors, seizures, and coma. Platelet and leukocyte counts may be depressed, and the odor of the urine has been described as similar to that of sweaty feet (100). Cerebral edema is present, and seizures are most often focal motor or generalized tonic. The initial EEG may be normal, but later studies may show deterioration, including dysmature features during sleep (25).
The symptoms of propionic acidemia also appear during the neonatal period, with seizures as the first symptom in a fifth of patients. Characteristic features include vomiting, lethargy, ketosis, neutropenia, periodic thrombocytopenia, hypogammaglobulinemia, developmental retardation, and intolerance to protein. Patients may have very puffy cheeks and an exaggerated Cupid’s bow-shaped upper lip. Convulsions are the rule, although more limited focal seizures have also been reported. The EEG shows background disorganization with marked frontotemporal and occipital slow-wave activity (25). In 40% of affected children, myoclonic seizures develop in later infancy, and older children may have atypical absence seizures.
Methylmalonicacidemia is the metabolic signature of seven biochemically distinct entities, all of which show decreased activity of methylmalonyl-CoA mutase. Stomatitis, glossitis, developmental delay, failure to thrive, and seizures are the major features, and lesions of the globus pallidus, visible on computed tomography or magnetic resonance images, are the characteristics. Diffuse tonic seizures and focal seizures with secondary generalization are the most frequent seizure types. Seizures may also be characterized by eyelid clonus with simultaneous upward deviation of the eyes. As in many other epileptogenic encephalopathies, the described EEG findings are multifocal spike discharges, depressed or slowed background activity, and sometimes lack of sleep spindles (25).
Pyruvate Dehydrogenase Deficiency, Pyruvate Carboxylase Deficiency, and Leigh Syndrome
Pyruvate dehydrogenase complex (PDC) is a multienzyme conglomeration of six subunits: pyruvate dehydrogenase (E1), dihydrolipoyl acetyltransferase (E2), dihydrolipoyl dehydrogenase (E3), an E3-binding protein (E3BP), pyruvate dehydrogenase kinase (PDK), and pyruvate dehydrogenase phosphatase (PDP) (5,6). The latter two components serve regulatory function. A majority of the mutations affect the E1 alpha subunit of the PDC, which impairs either stability or enzyme efficiency. Epilepsy may be due to a combination of structural abnormalities and energy failure, leading to clastic lesions (26,27).
There are various clinical manifestations of pyruvate dehydrogenase deficiency, ranging from acute lactic acidosis in infancy with severe neurological impairment to a slowly progressive neurodegenerative disorder (27,28). It is one of the most common defined genetic defects of mitochondrial energy metabolism. Structural abnormalities, such as agenesis of the corpus callosum, can be visible on neuroimaging. Seizures frequently occur and may take the form of infantile spasms and myoclonic seizures. In addition to hypsarhythmia, reported electrographic findings include multifocal slow spike-and-wave discharges. Treatment with the ketogenic diet helps with the paroxysmal aspects of the disorder.
Three clinical types of pyruvate carboxylase deficiency are recognized: an infantile form (type A), a severe neonatal form (type B), and an intermittent/benign form (type C). The neonatal type manifests with severe lactic academia, hepatomegaly, pyramidal tract signs, and death in the first few months of life. The infantile form is usually lethal in infancy or early childhood and begins in the first 6 months of life with episodes of lactic acidemia precipitated by an infection. Individuals with the intermittent form have normal or mildly delayed neurologic development and episodic metabolic acidosis. Seizures in the more severe forms are related to the hypoglycemia, which occurs secondary to dysfunction of the Krebs cycle. Hypsarhythmia, with or without infantile spasms, may be seen (29). Treatment with ACTH or the ketogenic diet can be very hazardous (30,31).
Leigh syndrome (subacute necrotizing encephalomyelopathy) may be caused by various metabolic defects, including cytochrome c oxidase deficiency, and defects in other enzymes involved in energy metabolism. (In the discussion section of his case presentation, Leigh noted that similar pathological changes could be seen in immature animals that were completely deprived of nourishment) (32). Mutations in various mitochondrial respiratory chain complexes (I, II, and V) as well as any of the three catalytic subunits (E1, E2, or E3) of the PDC may result in the same phenotype (33). In addition, rare causes of Leigh syndrome include myoclonus epilepsy with ragged red fibers (MERRF), mitochondrial encephalomyopathy with lactic acidosis and stroke-like symptoms (MELAS), and mitochondrial DNA depletion. Within the first year of life, classic features include failure to thrive, lactic acidosis, and developmental delay. As the disease progresses, infants develop spasticity, abnormalities of eye movements, and central respiratory failure. A variety of seizures may occur in Leigh syndrome, including focal and generalized (34). Cases of infantile spasms and hypsarhythmia (35) and several cases of epilepsia partialis continua (36) have also been reported. As is the case with the vast majority of epileptogenic encephalopathies associated with IEMs, EEG abnormalities, although present, do not appear to be distinctive enough to allow identification of the syndrome on electrographic criteria alone (37).
Disorders of Biotin Metabolism
Early Onset Multiple Carboxylase Deficiency (Holocarboxylase Synthetase Deficiency)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


