Table 42.1 Dana Point Clinical Classification of Pulmonary Hypertension (2008)1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Table 42.2 Prevalence of PH Groups | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||
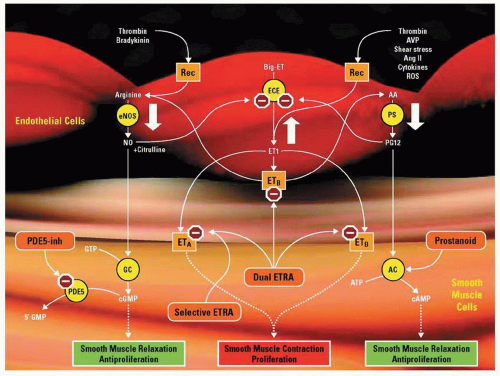 Figure 42.1 Three major mechanistic pathways are known to be perturbed in patients with PAH. (1) The NO pathway: NO is created in endothelial cells by type III NO synthase (eNOS), which in turn induces guanylate cyclase (GC) to convert guanosine triphosphate (GTP) to cGMP, a second messenger that constitutively maintains pulmonary artery smooth muscle cell (PASMC) relaxation and inhibition of PASMC proliferation. (2) The endothelin (ET) pathway: Big-ET (or pro-ET) is converted in endothelial cells to ET-1 (21 amino acids) by endothelin-converting enzyme (ECE). ET-1 binds to PASMC ETA and ETB receptors, which ultimately leads to PASMC contraction, proliferation, and hypertrophy. ET-1 also binds to endothelial cell ETB receptors. (3) The prostacyclin pathway: The production of PGI2 (prostacyclin) is catalyzed by prostacyclin synthase (PS) in endothelial cells. In PASMCs, PGI2 stimulates adenylate cyclase (AC), thus increasing production of cAMP from ATP, another second messenger that maintains PASMC relaxation and inhibition of PASMC proliferation. Importantly, the pathways interact as illustrated, modulating the effect of any single pathway. They also are impacted by transmitters and stimuli that act at cell membrane receptors (Rec). Examples of these include but are not limited to thrombin, bradykinin, arginine vasopressin (AVP), vessel-wall shear stress, angiotensin II (Ang II), cytokines, and reactive oxygen species (ROS). In addition, the effect of a transmitter depends on its specific site of action (such as PASMC ETA or ETB receptors versus endothelial cell ETB receptor). The large white arrows depict aberrations observed in these pathways among patients with PAH. The orange boxes represent agents with reported clinically beneficial effects in patients with PAH. PDE5-inh indicates PDE-5 inhibitor, e.g., sildenafil; ETRA, endothelin receptor antagonist, e.g., bosentan (dual), ambrisentan, and sitaxsentan (receptor A selective). Prostanoids, e.g., epoprostenol, treprostinil, and iloprost, supplement exogenously deficient levels of PGI2. Red stop signs signify an inhibitory effect of the depicted agents. Dotted arrows depict pathways with known and unknown intervening steps that are not shown.8 (Reproduced with permission from: McLaughlin VV, McGoon MD. Pulmonary arterial hypertension. Circulation 2006;114:1417-1431.) |
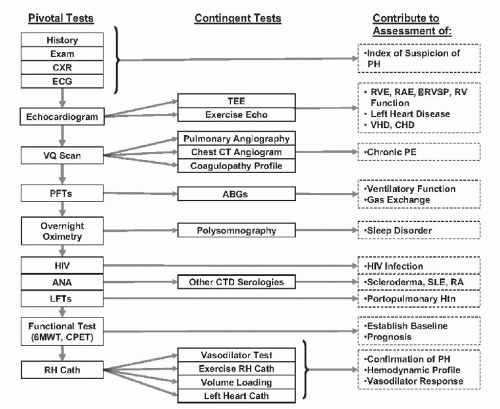 Figure 42.2 Diagnostic approach for suspected PAH. Pivotal tests are essential to establishing the definitive diagnosis of PAH.19 (Reproduced with permission from: McLaughlin VV, Archer SL, Badesch DB, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension. A report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians; American Thoracic Society, Inc.; and the Pulmonary Hypertension Association. Circulation 2009;119:2250-2294.) |
generally obtained early in the evaluation of patients with suspected PH. The tricuspid regurgitant jet is used to estimate the RVSP, and pressures >40 mmHg are consistent with PH. However, TTE can both under- and overestimate pulmonary artery pressures, as compared to the gold standard of RHC. In addition to the pressure measurements, TTE is useful in identifying elevated right-sided pressures by showing evidence of right atrial or ventricular enlargement, flattening of the interventricular septum, or an underfilled LV.
Ventilation/perfusion (V/Q) scan: the test of choice for the initial evaluation of chronic, surgically accessible thromboembolic disease. A PE protocol CT is excellent for identifying acute PE; however, is not sensitive for chronic thromboembolic disease. If indicated, pulmonary angiography can often be scheduled immediately following RHC if chronic thromboembolic disease is highly suspected.
Pulmonary function tests (PFTs): may show only mild restrictive disease and a mildly decreased diffusing capacity for carbon monoxide (DLCO); scleroderma patients may have a more marked decrease in DLCO.
Polysomnogram: should be performed if a patient endorses symptoms of sleep disordered breathing.
Blood tests: given the association between PH and both connective tissue disorders and chronic liver disease, antinuclear antibody (ANA) tests and liver function tests (LFTs) are routinely performed. HIV testing should be performed if a patient endorses risk factors. A complete blood count is helpful in identifying anemia as a cause of high-output heart failure. Thyroid function studies should be performed in all patients.
Six minute hall walk (6MWT): commonly used to objectively assess functional capacity; this test is often performed at baseline and repeated periodically to assess response to treatment; it is frequently used as an endpoint in clinical PAH trials.
with CCBs. Acute vasodilator testing should be avoided in patients with significantly elevated left heart filling pressures or low cardiac output. Veno-occlusive disease and pulmonary capillary hemangiomatosis should be considered in patients who experience pulmonary edema during vasodilator testing.22
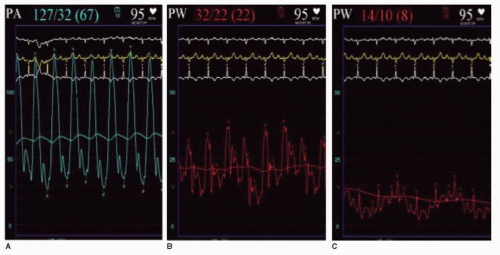 Figure 42.3 Pressure tracings depicting a PA waveform (A), underwedged PCWP with morphology intermediate between PCWP and PA waveforms (B), and a true PCWP waveform (C). |
Table 42.3 PAH-Specific Therapies | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 As needed to treat symptoms (may be needed for air travel)
As needed to treat symptoms (may be needed for air travel) Improved survival in observational studies involving patients with IPAH
Improved survival in observational studies involving patients with IPAH warfarin
warfarin Also recommended in advanced associated PAH
Also recommended in advanced associated PAH INR 1.5 to 2.5
INR 1.5 to 2.5 As needed to treat symptoms
As needed to treat symptoms furosemide
furosemide Indicated only in patients with positive vasodilator response during RHC; empiric administration without hemodynamic guidance may result in rapid deterioration
Indicated only in patients with positive vasodilator response during RHC; empiric administration without hemodynamic guidance may result in rapid deterioration diltiazem
diltiazem nifedipine
nifedipine amlodipine
amlodipine Avoid verapamil (decreased inotropy)
Avoid verapamil (decreased inotropy) Mechanism: replenishes PGI2, resulting in vasodilation, inhibition of platelet aggregation and inhibition of vascular SMCs
Mechanism: replenishes PGI2, resulting in vasodilation, inhibition of platelet aggregation and inhibition of vascular SMCs Flolan
Flolan Veletri
Veletri Indication: WHO class III to IV symptoms (IPAH or scleroderma spectrum of PAH)
Indication: WHO class III to IV symptoms (IPAH or scleroderma spectrum of PAH) Administration: continuous infusion requires a central venous catheter
Administration: continuous infusion requires a central venous catheter Overdose can result in high-output heart failure
Overdose can result in high-output heart failure Mechanism: same as epoprostenol
Mechanism: same as epoprostenol Remodulin
Remodulin Indication: WHO class II to IV symptoms (PAH)
Indication: WHO class II to IV symptoms (PAH) Tyvaso
Tyvaso Administration: continuous infusion via central venous catheter or subcutaneous sites (Remodulin), intermittent inhalation (Tyvaso)
Administration: continuous infusion via central venous catheter or subcutaneous sites (Remodulin), intermittent inhalation (Tyvaso) Mechanism: same as epoprostenol
Mechanism: same as epoprostenol Ventavis
Ventavis Indication: WHO class III to IV symptoms (PAH)
Indication: WHO class III to IV symptoms (PAH) Mechanism: nonselectively blocks vasoconstrictive and SMC mitogenic effects of endothelin-1 (ETA and ETB)
Mechanism: nonselectively blocks vasoconstrictive and SMC mitogenic effects of endothelin-1 (ETA and ETB) bosentan (Tracleer)
bosentan (Tracleer) Indication: WHO class II to IV symptoms (PAH)
Indication: WHO class II to IV symptoms (PAH) Administration: oral, 125 mg po BID
Administration: oral, 125 mg po BID Considerations: must monitor LFTs and hemoglobin; potential teratogen; may reduce sperm count
Considerations: must monitor LFTs and hemoglobin; potential teratogen; may reduce sperm count Mechanism: blocks vasoconstrictive and SMC mitogenic effects of endothelin-1 by selectively antagonizing ETA receptor (allows NO production via ETB receptor activation)
Mechanism: blocks vasoconstrictive and SMC mitogenic effects of endothelin-1 by selectively antagonizing ETA receptor (allows NO production via ETB receptor activation) ambrisentan (Letairis)
ambrisentan (Letairis) Indication: WHO class II to III symptoms (PAH)
Indication: WHO class II to III symptoms (PAH) Administration: oral, 5 or 10 mg once-daily
Administration: oral, 5 or 10 mg once-daily Considerations: must monitor LFTs and hemoglobin, potential teratogen, may reduce sperm count
Considerations: must monitor LFTs and hemoglobin, potential teratogen, may reduce sperm count Mechanism: prevents degradation of cGMP, resulting in vasorelaxation
Mechanism: prevents degradation of cGMP, resulting in vasorelaxation sildenafil (Revatio)
sildenafil (Revatio) Indication: WHO group I symptoms (PAH)
Indication: WHO group I symptoms (PAH) Administration: oral, 20 mg po TID
Administration: oral, 20 mg po TID Considerations: contraindicated in patients taking nitrates, may be used in conjunction with IV epoprostenol
Considerations: contraindicated in patients taking nitrates, may be used in conjunction with IV epoprostenol Mechanism: prevents degradation of cGMP, resulting in vasorelaxation
Mechanism: prevents degradation of cGMP, resulting in vasorelaxation Tadalafil (Adcirca)
Tadalafil (Adcirca) Indication: WHO group I symptoms (PAH)
Indication: WHO group I symptoms (PAH) Administration: oral, 40 mg po daily
Administration: oral, 40 mg po daily Considerations: contraindicated in patients taking nitrates, may be used in conjunction with IV epoprostenol
Considerations: contraindicated in patients taking nitrates, may be used in conjunction with IV epoprostenolStay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


