this is clinically obvious based upon medical history, electrocardiography, and echocardiogram.1 Because coronary disease is so common, and because it represents a potentially reversible cause of HF (if viable myocardium is present), cardiac catheterization including coronary angiography is recommended for most patients with new-onset HFrEF. Noninvasive assessment of ischemic heart disease is advocated by some but can be misleading with both false positives and false negatives.2 Once coronary disease has been excluded, the differential diagnosis includes the various causes of dilated cardiomyopathy (DCM; Table 43.1). The noninvasive clinical assessment may suggest a specific diagnosis such as sarcoidosis or Chagas disease, but in most instances the cause will remain undefined (i.e., idiopathic). Most cases of idiopathic cardiomyopathy likely represent the sequelae of prior myocarditis4 or genetic mutations.5 Indeed, a recent study reported that ∽25% of patients with “apparent” DCM had mutations within the titin gene.6 Only a few etiologies of DCM have pathognomonic histologic findings, but endomyocardial biopsy may be helpful in confirming or excluding those diseases. In 1230 patients who underwent endomyocardial biopsy at the Johns Hopkins Hospital (Baltimore, MD) for unexplained HFrEF, a specific cause was eventually determined in 50% of the patients using the results of the endomyocardial biopsy in combination with clinical information, although only 15% had a specific histologic diagnosis3, but using the results of the endomyocardial biopsy in combination with clinical information, a specific cause was eventually determined in 50% of the patients. In addition to coronary angiography and biopsy, ventriculography allows assessment of mitral regurgitation and dyskinesis, both of which can be targeted surgically. Ventriculography is used predominantly when other imaging techniques such as echocardiography or MRI are inadequate or inconsistent with the clinical examination.
Table 43.1 Causes of Dilated Cardiomyopathies (in the Order of Decreasing Frequency) | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||
Table 43.2 Serial Hemodynamics in Chronic Heart Failure Owing to an Idiopathic Dilated Cardiomyopathy | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
pressure (usually 50% of the PA systolic) as well as with the PA diastolic pressure (PAD; usually within 1 to 2 mmHg of PCWP) as long as pulmonary vascular resistance is <2 Wood units.17 Right ventricular pressure can also be used to estimate the PAD, since right ventricular pressure closely approximates pulmonary end-diastolic pressure at the time of pulmonary valve opening21 (maximal right ventricular dP/dt; Figure 43.2).
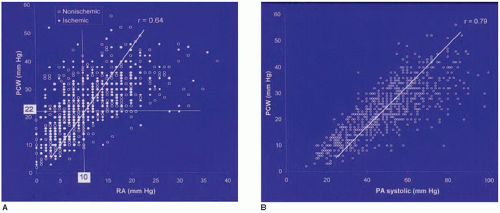 Figure 43.1 The relationship between (A) right atrial (RA) and pulmonary capillary wedge (PCW) pressure and (B) pulmonary capillary wedge pressure and pulmonary artery (PA) systolic pressure in 1,000 consecutive patients with chronic heart failure. (Reproduced with permission from Drazner MH, et al. Relationship between right and left-sided filling pressures in 1000 patients with advanced heart failure. J Heart Lung Transplant 1999;18:1126.) |
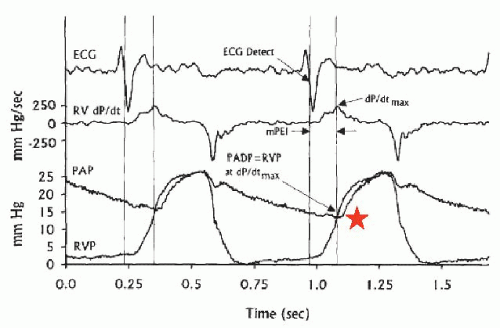 Figure 43.2 The pulmonary artery diastolic (PAD) pressure can be estimated from the right ventricular pressure (RVP) tracing since PAD is equal to right ventricular pressure at the maximal dP/dt (see asterisk). (Reproduced with permission from Reynolds DW, et al. Measurement of pulmonary artery diastolic pressure from the right ventricle. J Am Coll Cardiol 1995;25:1176.) |
entirely passive (i.e., owing simply to elevated PCWP, often termed “precapillary”) or owing to a combination of passive effects (e.g., elevated PCWP) and high pulmonary vascular resistance (i.e., >2.5 to 3 WU; often termed “reactive” or “postcapillary” PH). Recent data have shown that the presence of any type of PH is associated with increased risk of death or HF hospitalization in HFrEF, but “reactive” PH in particular is associated with a grim prognosis.22 Currently PH in HFrEF is not a specific therapeutic target, but recent studies using phosphodiesterase inhibitors have shown improvement in hemodynamics and quality of life, and multicenter clinical trials of pulmonary-specific vasodilators are underway.23
 Figure 43.3 Hemodynamic findings in decompensated heart failure from dilated cardiomyopathy. (See text for discussion.) |
contractility (Starling’s law). Severe left ventricular dysfunction can be well tolerated with normal or near-normal cardiac filling pressures, concomitant satisfactory functional capacity comparable to that of transplantation,26 and reasonable long-term survival. In fact, a low ejection fraction alone is not an indication for cardiac transplantation for morbidity or mortality reasons, and many patients with low EF are asymptomatic.27 In patients with HFrEF, stroke volume, stroke work index, and cardiac output can be maximized at pulmonary capillary wedge pressures as low as 10 mmHg (Figure 43.4).28 There is no justification for insufficient diuresis and dyspnea resulting from inadequate lowering of filling pressures, although recent data suggest that half of the patients with decompensated HF lose no weight or even gain weight during hospitalization for diuresis.16 In 754 consecutive patients with chronic heart failure referred electively for cardiac transplantation, tailored doses of vasodilators and diuretics increased the cardiac index from 2.1 ± 0.7 to 2.6 ± 0.6 L/minute per m2 despite a fall in the pulmonary capillary wedge pressure from 25 ± 9 to 16 ± 6 mmHg. In part, this improvement in forward cardiac output despite significant reduction of filling pressures is due to a decrease in mitral regurgitation caused by favorable changes in ventricular, atrial, and mitral annular geometry. This change can cause an increased forward stroke volume rather than an increase in total stroke volume.29,30 Left ventricular myocardial systolic performance can also improve owing to elimination of right ventricular volume overload with diuretics or nitrates,31,32 whereas myocardial diastolic performance can improve owing to reduced myocardial turgor that follows decreased venous congestion.33
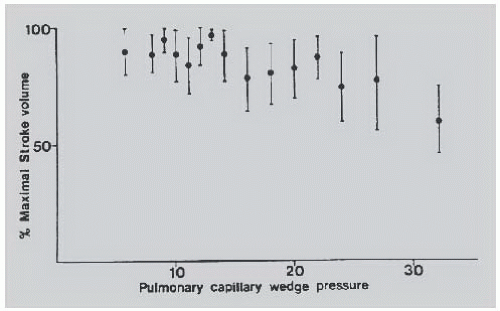 Figure 43.4 Maximal stroke volumes can be achieved at low filling pressures in advanced heart failure from dilated cardiomyopathy. (Reproduced with permission from Stevenson LW, et al. Maintenance of cardiac output with normal filling pressures in patients with dilated heart failure. Circulation 1986;74:1303.) |
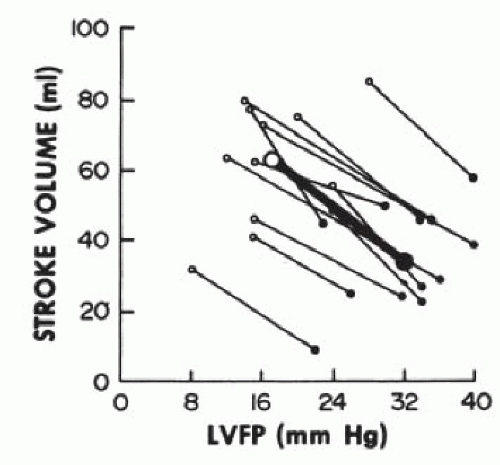 Figure 43.5 The effects of acute administration of nitroprusside on left ventricular filling pressures (LVFP) and stroke volume in patients with advanced heart failure. Maximal stroke volume is preserved at pulmonary capillary wedge pressures as low as 10 mmHg. (Reproduced with permission from Guiha NH, et al. Treatment of refractory heart failure with infusion of nitroprusside. N Engl J Med 1974;291:587.) |
to tailor their hemodynamics with vasodilators and diuretics. Such patients in general will be bridged to more definitive treatments such as mechanical support or cardiac transplantation. Inotropic agents, such as phosphodiesterase 3 inhibitors (e.g., milrinone) or beta agonists (e.g., dobutamine), will increase contractility (+dP/dt) and also decrease early and late diastolic pressures through their lusitropic (−dP/dt) effects42 (Figure 43.7; see also Chapter 28). Milrinone is also a vasodilator, which can augment the cardiac output over and above its direct inotropic action.43 Unfortunately these agents also increase myocardial oxygen demand, decrease LV efficiency, and have well-documented proarrhythmic effects.
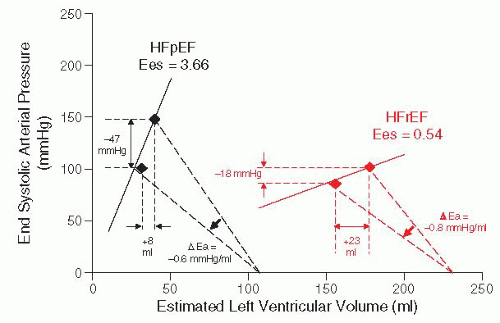 Figure 43.6 Effects of nitroprusside (SNP) on ventricular hemodynamics in HFpEF and HFrEF. In HFrEF, contractile function is severely depressed, resulting in a shallow end-systolic pressure-volume relation-ship (ESPVR, red straight line). The slope of the ESPVR is end systolic elastance (Ees). The shallow ESPVR in HFrEF explains how acute vasodilation with SNP (reduction in arterial elastance, Ea) results in small drop in blood pressure (−18 mmHg) and large enhancement in stroke volume (+23 mL). Conversely, HFpEF is characterized by a steep ESPVR (black solid line), and similar dosage and extent of vasodilation with SNP result in much more dramatic drop in blood pressure with less enhancement in stroke volume and cardiac output. (Reproduced with permission from Schwartzenberg, et al. Effects of vasodilation in heart failure with preserved or reduced ejection fraction: Implications of distinct pathophysiologies on response to therapy. J Am Coll Cardiol 2012;59:442-451.) |
stress increase even if myocardial mass is unchanged.48 In general, mild impairment of ventricular functional reserve is present, but it is demonstrable only with maximal exercise stress and may be in part a result of decreases in coronary flow reserve.54 The primary limitation in maximal cardiac output is owing to the denervated heart having a blunted heart rate response, and the Frank-Starling mechanism is exhausted early in the response to supine55,56 and upright exercise.57 This is partially offset by an increased sympathetic sensitivity, with dependence on circulating catecholamines for an adequate but delayed chronotropic exercise response.58
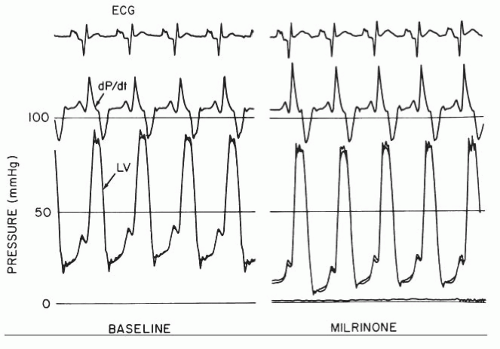 Figure 43.7 The hemodynamic effects of an inotropic agent, milrinone, in dilated cardiomyopathy. Milrinone increases contractility (positive dP/dt), improves lusitropy (negative dP/dt), and lowers preload (decreased left ventricular end-diastolic pressure [LVEDP]). The improvement in systolic and diastolic function occurs without an increase in systolic blood pressure. (Reproduced with permission from Baim DS, et al. Evaluation of a new bipyridine inotropic agent—milrinone—in patients with severe congestive heart failure. N Engl J Med 1983; 309:748.) |
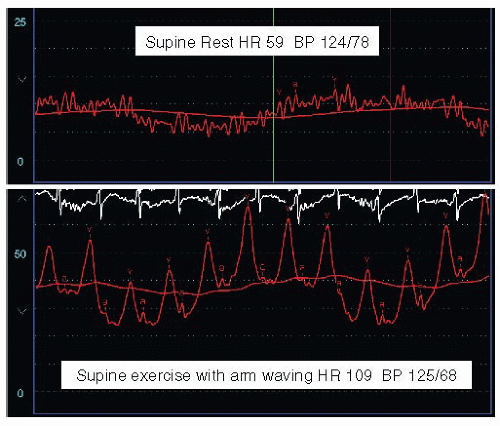 Figure 43.8 Effects of exercise on hemodynamics in dilated cardiomyopathy. Note the development of prominent V waves and dramatic increase of pulmonary capillary wedge pressure with exercise. This effect was achieved with supine arm exercise (moving bags of saline up and down in supine position) resulting in an almost twofold increase in heart rate (HR). The patient had an ejection fraction of 20%, an end-diastolic dimension of 7 cm, and moderately severe mitral regurgitation at rest. The lack of V waves and normal wedge pressure at rest are indicative of the chronic nature of the regurgitation. BP, blood pressure. |
and delayed increases in heart rate in response to physiologic stress. Afferent denervation results in dysregulation of sodium and water homeostasis as well as in abnormalities in peripheral vascular responses.59
Table 43.3 Resting and Exercise Hemodynamics After Cardiac Transplantation | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||
Table 43.4 Changes in Pulmonary Vascular Resistance to Various Maneuvers | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||
failure and is inversely correlated with oxygen consumption in these patients.64,65 The right ventricular (RV) response to chronic afterload increase is even more important than the load itself. Indeed, exercise capacity is better predicted by RV ejection fraction than by the left ventricular ejection fraction or pulmonary vascular resistance, reflecting the importance of the interaction between the pulmonary vasculature and right heart in limiting exercise capacity in heart failure.66
filling pressures are elevated, the increase in pulmonary blood flow can result in further increases in left ventricular filling pressure in a noncompliant ventricle and may produce alveolar pulmonary edema.83 However, in some patients there may be acute elevation in PCWP that remains asymptomatic.84 Adenosine85 and prostaglandin E186 produce similar effects. For these reasons, we generally avoid pulmonary-specific vasodilators for acute vasoreactivity testing when the PCWP or LVEDP is >20 to 25 mmHg and instead use nitroprusside, nitroglycerin, or milrinone. Table 43.5 summarizes the use of various acute vasodilators to assess reversibility of pulmonary hypertension in the catheterization lab. Endothelin antagonists also selectively vasodilate the pulmonary circulation87 and have been used in primary forms of pulmonary hypertension when left ventricular function is normal.88 However, oral endothelin antagonists in heart failure do not appear to be beneficial in clinical trials, and these agents have not been used for acute testing of pulmonary vasoreactivity.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


