Chapter 52 Proarrhythmia Syndromes
Proarrhythmia usually is defined as provocation of a new arrhythmia or aggravation of an existing arrhythmia during therapy with a drug at concentrations usually not considered toxic.1–9 However, drug-drug interactions that may lead to unrecognized elevations in drug concentrations, for example, an IKr (delayed rectifier potassium [K+] current)–blocking drug and a macrolide antibiotic, that can precipitate proarrhythmia. Other therapies (e.g., devices or ablation procedures) or pathophysiological conditions may also cause proarrhythmia.2,10,11 The various proarrhythmia syndromes are summarized in Box 52-1, and the drugs and conditions most frequently associated with proarrhythmia are listed in Table 52-1. Most types of proarrhythmia occur in the setting of structural heart disease, but proarrhythmia may also occur in individuals without apparent heart disease. Antiarrhythmic drugs have a higher risk of causing ventricular proarrhythmia, but drugs that are considered generally safe may cause proarrhythmia in susceptible individuals.2,4–6
Table 52-1 Drugs and Conditions Associated with Torsades de Pointes Ventricular Tachycardia
| DRUG CLASS | SPECIFIC DRUGS |
|---|---|
| ANTIARRHYTHMIC | |
| Class Ia | Quinidine |
| Procainamide | |
| Disopyramide | |
| Class Ic | Propafenone |
| Class III | Amiodarone |
| d, l-Sotalol | |
| d-Sotalol | |
| Dofetilide | |
| Ibutilide | |
| Azimalide | |
| Psychotropic | Haloperidol |
| Phenothiazines | |
| Risperidone | |
| Tricyclic antidepressants | |
| Tetracyclic antidepressants | |
| Diuretics | Furosemide |
| Hydrochlorothiazide | |
| Indapamide | |
| Metolazone | |
| Antimicrobial | Erythromycin |
| Clarithromycin | |
| Ciprofloxacin | |
| Levofloxacin | |
| Moxifloxacin | |
| Ofloxacin | |
| Trimethoprim-sulfamethoxazole | |
| Pentamidine | |
| Antifungal | Ketoconazole |
| Fluconazole | |
| Itraconazole | |
| Antihistamines | Diphenhydramine |
| Terfenadine* | |
| Astemizole* | |
| Cholinergic antagonists | Cisapride* |
| Narcotics | Methadone |
| DRUGS FOR OTHER CONDITIONS | |
| Bradycardia | Complete heart block or significant bradycardia |
| Electrolyte abnormalities | Hypokalemia |
| Hypomagnesemia | |
| Hypocalcemia | |
| Nervous system injury | Subarachnoid hemorrhage |
* No longer commercially available.
Drug-Induced Long QT Syndrome and Torsades De Pointes Ventricular Tachycardia
The greatest risk of ventricular proarrhythmia has been identified with drugs that prolong the Q-T interval on the electrocardiogram (ECG), which can predispose to a potentially life-threatening form of polymorphic ventricular tachycardia (VT), termed torsades de pointes VT (Figure 52-1).1–9 Other pathophysiological conditions that result in excessive prolongation of the Q-T interval may also cause this proarrhythmia (see Table 52-1).1–9 Syncope occurring early after the initiation of quinidine therapy was recognized as early as the 1920s, but it was not until the advent of continuous electrocardiographic monitoring that “quinidine syncope” was recognized to be caused by this pause-dependent polymorphic VT.12 The term torsades de pointes was initially coined by Dessertenne in 1966 to describe this polymorphic tachyarrhythmia that is often characterized by beat-to-beat changes in the QRS axis.13 Torsades de pointes VT often terminates spontaneously and may cause syncope, but it may transition into a sustained polymorphic VT or ventricular fibrillation (VF) and cause sudden cardiac death (SCD) (see Figure 52-1).
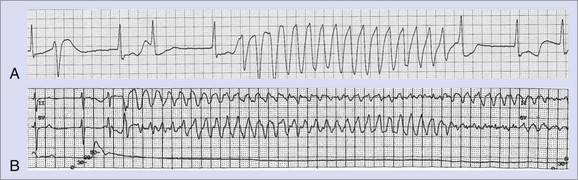
The most common drugs and conditions associated with excessive Q-T interval prolongation and torsades de pointes VT are summarized in Table 52-1. An up-to-date list of drugs associated with torsades de pointes VT is maintained at www.longqt.org. Antiarrhythmic drugs that prolong the ventricular action potential duration (APD), including class Ia drugs (quinidine, procainamide, disopyramide) and class III drugs (amiodarone, sotalol, dofetilide, ibutilide, azimilide), have all been reported to cause torsades de pointes VT.2–9 Rarely, the class Ic drug propafenone has been reported to cause torsades de pointes VT. Associated bradycardia and electrolyte abnormalities (hypokalemia, hypomagnesemia, or both), often caused by diuretic use, increase the probability of torsades de pointes VT in the setting of class Ia or III antiarrhythmic drug use. Although amiodarone significantly prolongs the Q-T interval, the incidence of torsades de pointes VT associated with amiodarone use is relatively low.2,14 Diuretics, by virtue of causing profound hypokalemia or hypomagnesemia, may be associated with torsades de pointes VT when other drugs are not used. The diuretic indapamide, which blocks the slowly activating component of the slow delayed rectifier K+ current (Iks), may cause excessive Q-T interval prolongation and torsades de pointes VT.15
Noncardiovascular drugs associated with torsades de pointes VT include tricyclic antidepressants (e.g., imipramine), antipsychotic drugs (e.g., haloperidol, risperidone, phenothiazine), antihistamines (e.g., diphenhydramine, terfenadine, astemizole), macrolide antibiotics (e.g., erythromycin, clarithromycin), quinolones (e.g., ciprofloxacin, levofloxacin, moxifloxacin, ofloxacin), pentamadine, anti-fungal agents, and cisapride.1–9 A number of drugs, including cisapride, terfenadine, and astemizole, have been withdrawn from the market because of an unacceptably high incidence of torsades de points VT. The risk of torsades de pointes VT caused by terfenadine or cisapride is attributed to the accumulation of these drugs in plasma as a consequence of coadministration of another drug (e.g., erythromycin or ketoconazole) that inhibits their cytochrome P450 3A4–mediated biotransformation to noncardioactive metabolites.5,6,8
Isolated profound bradycardia (most often in the setting of complete heart block) may be associated with profound Q-T interval prolongation and torsades de pointes VT, particularly in the setting of drugs or other factors that prolong the Q-T interval.13 Neurologic events such as subarachnoid hemorrhage have been associated with marked repolarization abnormalities, Q-T interval prolongation, and torsades de pointes VT. Adrenergic stimuli in the setting of dobutamine infusion have also been reported to cause torsades de pointes VT. Case reports have also implicated anesthetic agents, including halothane, as a cause for torsades de pointes VT.
Cellular Mechanisms of Torsades De Pointes Ventricular Tachycardia
An increase in inward currents, a reduction of outward currents, or a combination of both during phase 2 and 3 of the ventricular action potential leads to prolongation of the ventricular action potential, which manifests as Q-T interval prolongation on the ECG (Figure 52-2).1–7 Mutations of genes encoding K+, sodium (Na+), and calcium (Ca2+) channels have been linked to the congenital long QT syndrome (LQTS). Disease states such as left ventricular hypertrophy and heart failure are associated with a reduction in outward currents caused by the downregulation of transient outward K+ current (Ito), IKr, the inward rectifier K+ current (IK1), or all, which occurs in a spatially heterogeneous manner.16,17 A reduction in net outward currents, an increase in inward currents, or a combination of both, facilitates the development of early afterdepolarizations (EADs) that develop in M cells or Purkinje cells because of the activation or reactivation of arrhythmogenic inward currents, including Ca2+ channels or the Na+-Ca2+ exchange current.1–718 These EADs may initiate torsades de pointes VT, which is then maintained by re-entry (see Figure 52-2).1,6 Intracellular calcium overload, as in the setting of heart failure, may facilitate the development of EADs in the setting of acquired LQTS.17
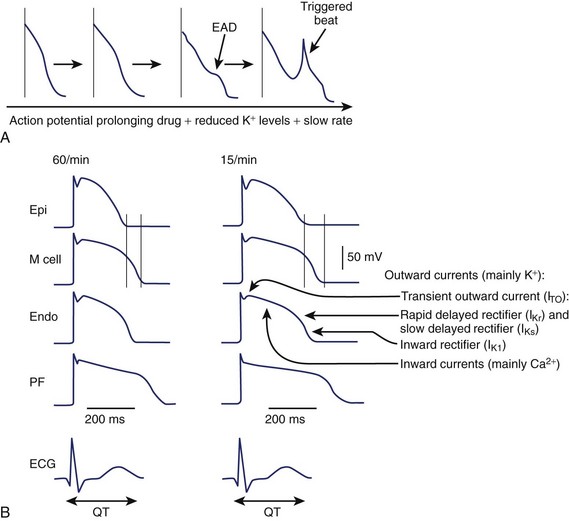
In the ventricular myocardium, the APD varies in a spatially heterogeneous manner because of differences in current densities in specific cell types (Purkinje fibers and endocardial, mid-myocardial, or epicardial cells).1,6,16,18 The ventricular APD is longest in the mid-myocardial layer (M cells) and shorter in the epicardial and endocardial regions. This dispersion of ventricular repolarization is increased in disease states, including ventricular hypertrophy, and may be further exaggerated by the administration of a drug that prolongs the Q-T interval.16 Marked spatial dispersion of ventricular repolarization appears to be a prerequisite for torsades de pointes VT because it provides the functional substrate for re-entry.19 Functional block may vary on a beat-to-beat basis, contributing to spiral re-entrant waves, which explains the polymorphic nature of the arrhythmia.1,6,20
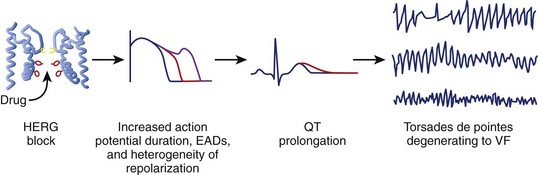
Although multiple ionic mechanisms may contribute to the development of torsades de pointes VT, the vast majority of drugs associated with torsades de pointes VT are thought to act by blockade of the rapid component of IKr (Figure 52-3).1,3,7 The HERG gene (also known as KCNH2) regulates the expression of IKr. The molecular structure of the drug-binding domain in the HERG channel makes it more vulnerable to blockade by a variety of drugs compared with other K+ channels. A small proportion of individuals who develop drug-induced torsades de pointes may have subclinical forms of congenital LQTS.3–621 Some polymorphisms of genes responsible for the expression or regulation of the ion channels involved in congenital LQTS have also been implicated in drug-induced torsades de pointes.3,4,21 Mutations in HERG may enhance blockade of the channel by certain drugs.
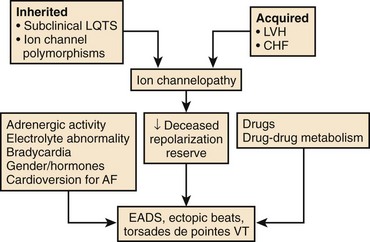
A normal QT at the start of antiarrhythmic drug therapy does not exclude the risk for proarrhythmia.2,5,7 The concept of reduced repolarization reserve and the risk of proarrhythmia was initially introduced by Roden.5,22 Cardiac repolarization is determined by IKr and IKs as well as other inward and outward currents during the plateau of the action potential. A defect in one of these currents may not be apparent if other currents that contribute to repolarization are intact. Roden has hypothesized that “physiologic processes such as drug metabolism or cardiac repolarization include multiple redundancies” and that “loss of these redundancies due to congenital or acquired conditions may enhance susceptibility to proarrhythmic responses even in the absence of a baseline phenotype.”5 For example, decreased expression of IKs because of a mutation in one of the genes responsible for expression of IKs, or because of a pathophysiological state such as heart failure or atrial fibrillation (AF), may not become apparent until the individual is treated with a drug that blocks IKr, which results in marked prolongation of the Q-T interval and proarrhythmia in this setting of reduced repolarization reserve (Figure 52-4).
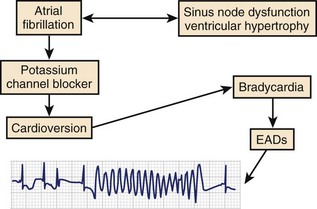
The period immediately after cardioversion to sinus rhythm from AF appears to be a time of great risk for torsades de pointes VT.9,23,24 The potential mechanism underlying this event is shown in Figure 52-5. The sustained tachycardia and the neurohumoral changes associated with AF may alter some electrophysiological properties, including downregulation of repolarizing currents. Restoration of sinus rhythm is often associated with bradycardia and action potential prolongation that could be further exaggerated by the downregulation of K+ channels. The action potential prolongation also facilitates increased sarcoplasmic calcium, predisposing to triggered activity. A similar situation may occur after atrioventricular (AV) junction ablation for the management of persistent AF. The early period after ablation is a particularly vulnerable period for SCD, which is believed to be caused by bradycardia-dependent polymorphic VT.25 Successful AV junction ablation is associated with abrupt slowing of the heart rate. Sudden slowing of the heart rate and the associated prolongation of the ventricular APD after ablation could increase the likelihood of EADs. Consistent with this hypothesis, we have observed exaggerated bradycardia-dependent prolongation of the Q-T interval and increased Q-T interval dispersion in patients with significant left ventricular dysfunction after total AV junction ablation.26
Risk Factors for Torsades de Pointes Ventricular Tachycardia
The risk factors for torsades de pointes VT are summarized in Box 52-2.1–9,27,28 The presence of multiple risk factors (e.g., female gender, cardiac hypertrophy, electrolyte abnormalities, and prior history of VT) dramatically increase the risk of developing torsades de pointes VT. Diuretic use in the setting of normal serum potassium concentrations has been reported to be a risk factor for torsades de pointes VT.29 This may be caused by total body potassium depletion that may not be reflected by serum potassium concentrations or the direct action potential–prolonging effects of some diuretic drugs.
Women have a twofold to threefold greater risk of developing torsades de pointes VT during treatment with action potential–prolonging drugs independent of other risk factors.27,30 In the general population, women have longer Q-T intervals compared with men. In experimental models, females have longer ventricular APDs and increased transmural dispersion of repolarization compared with males, which may be explained, in part, by reduced expression of IKr as well as other K+ currents.28 Testosterone shortens the ventricular APD, whereas estrogen prolongs the APD. Gender-specific differences in drug transport and metabolism may also contribute to the modulation of the pharmacokinetics of IKr-blocking drugs.28
The risk of torsades de pointes VT is generally related to drug dose.6,31 However, torsades de pointes VT has been described as an idiosyncratic reaction that occurs during quinidine therapy, and syncope secondary to torsades de pointes occurs within hours of initiation of this drug therapy.6,12 The development of torsades de pointes VT at low quinidine concentrations may be secondary to the IKr-blocking effects in conjunction with augmentation of the late Na+ current resulting in a decrease in net repolarizing currents; however, at higher quinidine concentrations, the more dominant Na+ channel–blocking effects balance the IKr block and restore the repolarization reserve.6 The risk of torsades de pointes VT associated with sotalol or dofetilide therapy clearly increases significantly with dose, which is further exacerbated by other risk factors, including renal failure.31 The incidence of ventricular proarrhythmia during sotalol initiation at doses up to 320 mg/day was 1.8%; this incidence increased to 4.5% at doses up to 480 mg/day and to 6.8% at doses greater than 640 mg/day.
Some cardiac disease processes such as ventricular hypertrophy or significant left ventricular dysfunction are associated with significant prolongation of the APD.32–34 These electrophysiological changes are caused by changes in repolarizing current densities, including decreases in Ito, IK1, IKr, and IKs.16,35 Changes in these repolarizing currents in their densities, balance, or both may alter the response of specific channel blockers, resulting in exaggerated prolongation of the APD and increased dispersion of ventricular repolarization that provides the substrate for torsades de pointes VT.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


