Chapter 18 Principles of Hemodynamics Applied to Cardiac Arrhythmias
The hemodynamic consequences of cardiac arrhythmias depend on various factors, including the filling and emptying capacity of the atrium and the ventricle, the duration and relative timing of systole and diastole, as well as neurohormonal influences on cardiac contractility and peripheral tone. Early studies examined these effects in various arrhythmias, including atrial fibrillation (AF), Wolff-Parkinson-White syndrome, and ventricular tachycardia (VT).1,2 This chapter expands on this pioneering work to provide a current framework for considering the important hemodynamic consequences of cardiac arrhythmias.
Atrial Mechanics
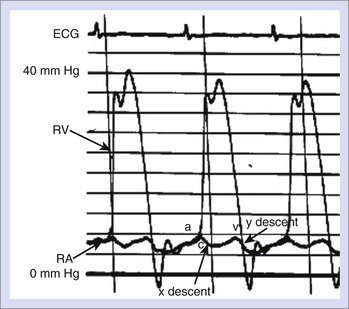
Pressure and flow recordings from the right and left atria under normal conditions consist of two major positive deflections: (1) the “a” wave and (2) the “v” wave (Figure 18-1). The a wave results from atrial contraction and is immediately followed by the “x” descent, representing atrial relaxation. A small “c” wave in the right atrial tracing occurs during the x descent and represents tricuspid valve closure. The atrial v wave occurs during ventricular contraction, which results in the descent of the tricuspid valve annulus and the passive filling of the atrium from the venous system. As the right ventricular pressure falls below that of the atrium, the tricuspid valve opens, and atrial blood is emptied into the ventricle, resulting in the “y” descent.
Early studies by Gesell and Wiggers estimated the contribution of atrial systole to overall cardiac output to be between 10% and 35%.3,4 Subsequent studies have shown that the atrial contribution to left ventricular (LV) performance operates via a Frank-Starling relationship, whereby cardiac output is augmented by an increase in end-diastolic volume (preload) of the ventricle.3 Loss of atrial systole results in a decrease in cardiac output by causing a downward shift of the Frank-Starling curve; that is, the smaller left ventricular end-diastolic diameter from diminished filling results in a smaller stroke volume. This relationship is independent of changes in systemic afterload.
Although less well studied than ventricular mechanics, left atrial (LA) pump function has been characterized in both normal and pathologic states.4 In the early stages of LV dysfunction, atrial contraction is augmented via an upregulation of β-myosin heavy chains in response to the increased afterload from worsening LV compliance. This improvement in LA mechanical work may aid in initial compensation of heart failure symptoms. As ventricular pressure and volume continue to increase, however, intrinsic atrial contractility deteriorates, and circulatory failure progresses. As heart failure progresses and, consequently, LV end-diastolic pressure increases, the capacity of atrial systole to augment cardiac output diminishes.
The reservoir function of the left atrium, which includes the passive, early-phase filling during atrial relaxation and the late-phase filling during ventricular systole, has been shown to be an independent determinant of cardiac output.5 Disease states that reduce atrial compliance, including conditions of chronic pressure overload such as mitral stenosis, hypertrophic cardiomyopathy, or advanced LV systolic dysfunction, may result in decreased cardiac output despite preserved atrial contraction. The loss of atrial systole or atrioventricular synchrony in the background of such diseases may result in the sudden deterioration of previously well-compensated cardiac function. The effects of specific atrial arrhythmias on atrial and ventricular hemodynamic functions are discussed below.
An important but perhaps under-recognized component of atrial mechanical function is the conduction between the right and left atria. The classic electrocardiographic criteria established for LA enlargement correlates with elevated pulmonary capillary wedge pressure only in patients with cardiomyopathy or rheumatic mitral valve disease.6 In patients without cardiomyopathy, the electrocardiogram pattern of LA enlargement may represent a prolongation of interatrial conduction time (IACT), often with normal LA size and pressures. Disease states such as AF, hypertension, and mitral regurgitation are associated with adverse atrial myocyte remodeling. Abnormalities in IACT are also common in these conditions, but whether remodeling leads to interatrial conduction delays or is a result of incoordinate atrial contraction is unknown. In patients undergoing cardiac resynchronization therapy for cardiomyopathy, those with an IACT greater than 100 ms have had larger LA volumes and lower indices of LA function at follow-up compared with patients with normal IACTs.7 The presence of severely prolonged IACT in this population has important implications for optimal programming of the atrioventricular delay as AV nodal conduction may be more rapid than right-to-left atrial conduction in up to 30% of patients.
Prolongation of the P-wave duration and IACT is common in patients with AF.8 Slowed conduction in the setting of increased atrial dimension increases the number of wavelets that coexist, thereby increasing the likelihood that AF will be sustained. In addition to prolongation of the normal right-to-left atrial route of conduction via Bachmann’s bundle, atrial fibrosis and remodeling often result in alternative conduction pathways to the LA, such as the margin of the fossa ovalis or coronary sinus ostial connections. Shortening of the IACT via bi-atrial epicardial pacing after coronary artery bypass grafting has been shown to decrease the incidence of AF as well as to improve pulmonary capillary wedge pressure and cardiac output. The complex interplay of atrial hemodynamics and geometry on IACT and susceptibility to arrhythmia warrants further study.
Premature Contraction
Closely coupled PACs result in incomplete ventricular filling and lower-than-usual left ventricular end-diastolic volume. Immediate postectopic beats, however, demonstrate augmented filling and, thus, increased stroke volume. In addition, postextrasystolic potentiation, which is the increased contractility seen in postectopic beats, may further enhance cardiac output. Hemodynamic studies have shown that in the majority of patients with PACs, cardiac output is not significantly changed.9 In those with more frequent PACs, however, cardiac output may decrease by as much 25%.
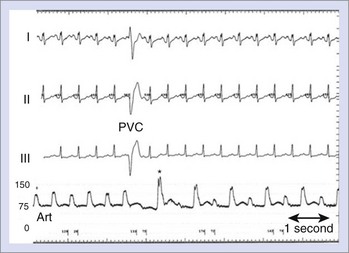
The effect of PVCs on LV stroke volume is often more profound than that of PACs. The shortened end-diastolic filling time as well as the compensatory pause before the postectopic beat can set up profound changes in LV contractility, such as those seen in pulsus alternans (Figure 18-2). Similar to the effect of PACs, the immediate effect of PVCs on arterial blood pressure is related to the coupling interval of the ectopic beat, with extremely short-coupled PVCs often resulting in no discernible arterial pressure. The site of origin of PVCs, on the other hand, does not appear to affect the relative cardiac output.10
Frequent PVCs can lead to deterioration of cardiac function in an otherwise structurally normal heart. Patients with the highest percentage of PVC beats (>20% of total daily beats) show evidence of reverse LV remodeling, reduced ejection fraction, and a greater amount of mitral regurgitation compared with those with infrequent PVCs. Over time, these patients may show evidence of progression of heart failure, with worsening New York Heart Association (NYHA) functional class, elevated brain natriuretic peptide, and elevated baseline heart rate (HR). The mechanism for LV dysfunction with frequent PVCs is likely multifactorial. In patients whose PVCs originate in the right ventricular outflow tract, the left bundle branch block (LBBB)–type beat results in septal dyskinesia as well as abnormal basal-to-apical ventricular activation. Cumulatively, these effects, over time, may lead to inefficient myocardial contraction, with progressive LV stiffening, elevated LV end-diastolic pressure, and ventricular dilation. Closely coupled PVCs cause calcium release, which results in postsystolic potentiation and improvement in ejection fraction in the beat immediately thereafter. In the long term, however, chronic calcium overload might result in cellular dysfunction and worsening of cardiac performance. The cause of LV dysfunction is likely unrelated to elevated HR, as mean HR has not been found to significantly affect the degree of ventricular dysfunction in patients with frequent PVCs.11
Reversal of adverse hemodynamic and ventricular remodeling effects of frequent PVCs is possible with the use of antiarrhythmic medications or catheter ablation targeting the PVC focus.12,13 A randomized, controlled study examining the use of class IC antiarrhythmic drugs to suppress asymptomatic or minimally symptomatic PVCs in patients after myocardial infarction was terminated early because of increased mortality in the treatment arm.14 The impact of these medications on mortality among patients without coronary disease has not been well studied.15 Studies of catheter ablation for frequent, monomorphic PVCs in patients with structurally normal hearts have shown the most common site of origin to be the outflow regions of the right ventricle (RV) and the LV or, more rarely, in the posteroseptal region of the LV chamber.16 Acute and midterm success rates for catheter ablation of these foci are excellent, although no long-term, prospective data are yet available. Recently, catheter ablation of post-infarction PVCs in patients with ischemic cardiomyopathy and greater than 5% daily PVC burden was shown by cardiac magnetic resonance imaging to be highly successful with an improvement of cardiac function to near-normal in patients without high-density scar.17 While these data demonstrate that elimination of the PVC focus results in reversal of LV dilation and improvement of ejection fraction (EF), no evidence that this has a beneficial impact on mortality has yet been presented.
Atrial Fibrillation
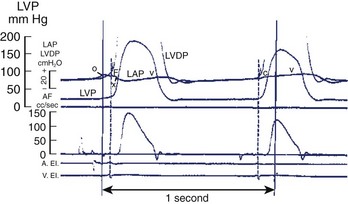
The atrial chamber serves as a conduit between the venous bed and the ventricles. More than that, however, atrial systole also contributes importantly to ventricular filling. Early animal studies found that atrial systole increased ventricular filling by roughly 35% and that the magnitude of the effect depended on both the strength of the atrial contraction as well as relative timing during ventricular diastole.18 In addition to augmenting forward ventricular stroke volume, atrial systole is also important in maintaining a low mean atrial pressure by allowing properly timed closure of the mitral valve during LV diastole (Figure 18-3). The contribution of atrial systole appears to be particularly important at higher heart rates.
Loss of Atrial Systole in Normal Hearts
The occurrence of AF can have significant hemodynamic consequences in structurally normal hearts. The irregularity of the rhythm, independent of HR, has been shown to decrease cardiac output in experimental models.19 The most likely explanation for this finding is that beat-to-beat changes in ventricular filling alter myocardial contractility via the Frank-Starling mechanism. The beats with longer R-R intervals do not entirely compensate for those beats with short R-R intervals, and an overall loss of cardiac output ensues. A second possible mechanism for this finding is that sudden changes in ventricular cycle length cause inefficient ventricular mechanics and wasted energy because of incomplete cellular restitution during an abbreviated ventricular diastole. In support of the independent deleterious effect of heart rate irregularity, a small trial of atrioventricular junction ablation in patients with chronic AF and a normal range of HR (60 to 100 beats/min) showed improvement in EF and heart failure symptoms at 1-year follow-up. In contrast, relative regularization of the R-R intervals using ventricular rate-smoothing algorithms in patients with AF and permanent pacemakers has failed to show significant symptomatic improvement.20
Incessant rapid ventricular rates during AF can lead to ventricular dysfunction and progressive congestive heart failure (CHF). Hemodynamic changes, characterized by markedly elevated ventricular filling pressures and impairment of LV contractility, are seen after as little as 24 hours of rapid ventricular pacing in animal models. As cardiac output diminishes, systemic vascular resistance is typically increased, and mitral regurgitation may develop as a consequence of increased afterload. Calcium overloading of myocardial cells during chronic tachycardia may also lead to impairment of LV relaxation and progressive diastolic dysfunction. With ongoing high-rate pacing, ventricular filling pressures continue to rise with a plateau at about 1 week. Cardiac output may continue to deteriorate for up to 5 weeks, with eventual development of symptoms of CHF.21
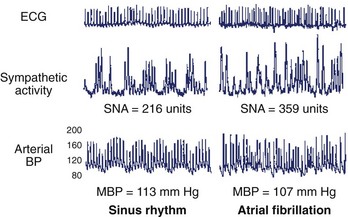
Neurohormonal activation during AF may have additional deleterious hemodynamic effects in patients with otherwise normal cardiac function. HR irregularity and sustained tachycardia lead to increased sympathetic nerve activity. This, in turn, may result in an increase in right atrial pressure and decreased systemic blood pressure because of cardiopulmonary baroreceptor activity (Figure 18-4).22 Prolonged neurohormonal activation with elevated levels of catecholamines and angiotensin II may promote long-term structural changes to the LA because of atrial fibrosis.23 In addition, atrial stretch from chronically elevated pressure is thought to activate cardiac mechano-receptors, which, in turn, leads to increased cardiac vagal activity. Vagotonic effects in the cardiac conducting system include a shortening of the atrial effective refractory period, which is conducive to the initiation and maintenance of AF.24 Interestingly, catheter ablation of the vagal nerve fibers during pulmonary vein isolation (PVI) may reduce the recurrence of AF after ablation, perhaps because of attenuation of these adverse parasympathetic effects (Figure 18-5).25
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


