Chapter 12 Principles of Clinical Pharmacology
Basic Concepts in Pharmacokinetics
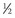 ). From this concept, the following equation is derived:
). From this concept, the following equation is derived: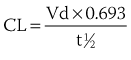
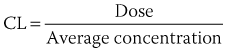
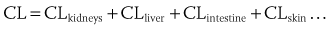
Absorption of drugs can vary with timing of dose in relation to a meal. For example, concentrations of dronedarone (a currently investigational antiarrhythmic drug) are twofold to threefold higher when taken with a meal compared with peak concentrations when taken on an empty stomach.1
Intersubject Variability in Drug Action
Several factors can modulate the response obtained following the administration of a particular drug to a particular patient at a particular time. This statement argues against the “one size fits all” concept and clearly defines the need for individualized drug therapy. To fully integrate the basic principles underlying clinical pharmacology, the prescriber needs to understand the principles of pharmacokinetics, pharmacodynamics, and drug efficacy. Figure 12-1 depicts the three major principles that define the relationship between drug dose and clinical outcome.
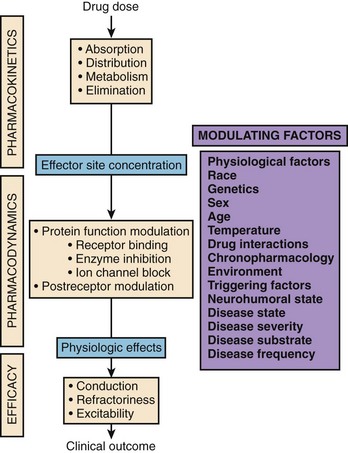
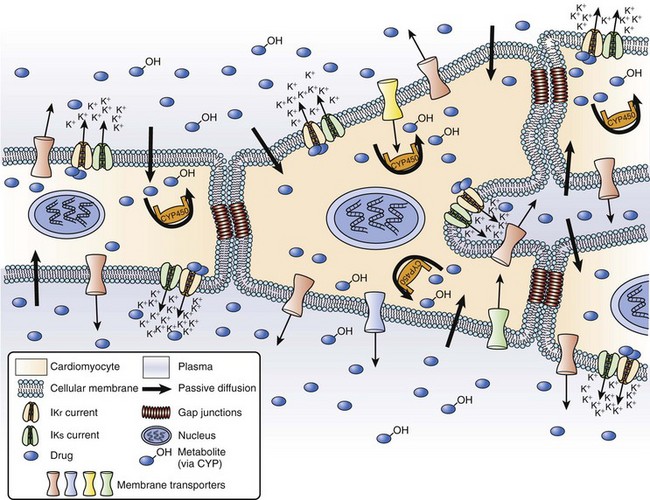
As discussed earlier, pharmacokinetics describes the relationship between the dose administered and the observed concentrations of a drug or its metabolites in selected biologic fluids. Concentrations of active or toxic substances at their effector or toxic sites are often of the greatest interest. Pharmacodynamics describes the relationship between the concentration of an active substance at its effector site and the physiological effects observed. Currently, most drugs are aimed at either direct or indirect modulation of a protein function. For most of them, there is a range of concentrations for which changes in protein function are linearly related to drug concentration. Finally, drug efficacy links the physiological effects observed following the administration of a drug to clinical outcome. Several major clinical trials in recent years, such as the Cardiac Arrhythmia Suppression Trial, have demonstrated that achievement of expected pharmacodynamic response is not necessarily related to a desirable clinical outcome (i.e., drug effectiveness).2,3
Drugs with a Narrow Therapeutic Index: Antiarrhythmic Agents
The notion that monitoring plasma drug concentrations could provide a method for adjusting doses to reduce inter-individual variability in response arose during the development of new antimalarial drugs during World War II. Shortly thereafter, this notion was applied to quinidine therapeutics.4 This concept was derived from the well-recognized relationships between “normal” plasma ion concentrations or hormonal levels and a “normal” physiological state. Using such a framework, it was observed in initial trials that plasma concentrations of quinidine below 3 µg/mL were rarely associated with an antiarrhythmic response, whereas concentrations above 8 µg/mL were frequently associated with QRS widening, cinchonism, and hypotension.5 Thus, a tentative therapeutic range of 3 to 8 µg/mL was defined.
Using the same approach, relatively well-defined therapeutic ranges were also established for lidocaine (4 to 8 µg/mL), mexiletine (500 to 1000 ng/mL), and procainamide (4 to 8 µg/mL) for patients presenting with ventricular arrhythmias.6–9 However, as drug assays developed further and experience accumulated, it became evident that the therapeutic concentration window was very wide with these antiarrhythmic agents and that wide intersubject variability existed. Therapeutic ranges, such as the one for quinidine (2 to 5 µg/mL), had to be redefined because of impurities and metabolites interfering with early fluorometric methods.10 Also, the overlap between effective and toxic concentrations (narrow therapeutic/toxic window) in different patients was significant, and it became almost impossible to predict, for a specific patient, plasma levels associated with efficacy or toxicity.
Subsequently, another important source of intersubject variability was identified in patients treated with the potent class Ic antiarrhythmic agent encainide.11 In a small clinical study, 10 of 11 patients with ventricular arrhythmias responded to the drug (encainide) with arrhythmia suppression and QRS widening, and the eleventh had no response. In the 10 responders, peak plasma encainide ranged from 3 to 200 ng/mL. In the single nonresponder, peak plasma encainide was the highest (300 ng/mL). Further studies demonstrated the importance of active metabolites (O-demethyl encainide [ODE] and 3 methoxy-O-demethyl encainide [MODE]) in accounting for encainide action, but a simple therapeutic range—based solely on the plasma concentrations of the parent compound or in combination with the metabolites—could not be defined.12
Propafenone is another class Ic antiarrhythmic agent that shows wide intersubject variability in its response and in the formation of active metabolites.13 In addition, the drug exhibits varying electrophysiological (sodium, calcium, and potassium channel block) and pharmacologic (β-blocking) effects depending on the route of administration, the metabolism status, and the plasma concentrations of its enantiomers.13,14 Several investigators have tried to derive combined therapeutic ranges for the metabolites—the enantiomers—and for the combinations of the parent drug plus metabolites, without success.
The situation with antiarrhythmic agents is not unique and is observed with other drugs that have a narrow therapeutic index. For example, doses and plasma concentrations of warfarin that were required to maintain the international normalized ratio (INR) within acceptable limits (2 to 3) vary widely among individuals.15–17 There is no rationale to use the plasma concentrations of each warfarin enantiomer, rather than INR values, to adjust warfarin doses.
Pharmacogenetics
Genetically Determined Pharmacokinetic Factors
Genetically determined abnormalities in the ability to biotransform drugs range from apparently benign conditions such as Gilbert’s syndrome (a deficiency in glucuronyl transferase activity) to the rare but potentially fatal syndrome of pseudo-cholinesterase deficiency. This most widely studied polymorphic drug oxidation trait is a deficiency in the cytochrome P450 isozyme (CYP2D6) responsible, among others, for the biotransformation of the antihypertensive drug debrisoquine to its inactive 4-hydroxy metabolite.18,19 Following the oral administration of a single 10-mg dose of debrisoquine, a metabolic ratio (debrisoquine/4-hydroxydebrisoquine), established from an 8-hour urinary excretion profile, can discriminate between two distinct phenotypes.20 Individuals with a ratio greater than 12.6 are defined as poor metabolizers (PMs), whereas a value less than this antimode reflects the ability to extensively metabolize (EM) the probe drug. Family studies indicated that the deficient trait is inherited as an autosomal recessive character.18 Regardless of geographic location, about 5% to 10% of whites are PMs. At the other end of the spectrum, 2% to 5% are known as ultra-rapid metabolizers (UM), since they exhibit very high expression levels and activity of CYP2D6.
The CYP2D6 gene is located on the long arm of chromosome 22 (q11.2-qter).21 Deletion or transition mutations in the gene lead to splicing errors during messenger ribonucleic acid (mRNA) processing and result in unstable proteins.22,23 Therefore, the CYP2D6 protein is functionally absent in PMs. Deoxyribonucleic acid (DNA) assays based on allele-specific amplification with the polymerase chain reaction (PCR) allow identification of approximately 95% of all PMs.23–25
CYP2D6 activity can also be inhibited by drugs, including quinidine, some tricyclic antidepressants, and some selective serotonin reuptake inhibitors (SSRIs; fluoxetine and paroxetine).26
CYP2D6 can metabolize substances via various C-oxidations, including aromatic, alicyclic, and aliphatic hydroxylation; N- and S-oxidation; as well as O-dealkylation. For example, the metabolism of several classes of cardiovascular drugs such as β-blockers and class I antiarrhythmic drugs, as well as the metabolism of neuroleptics and antidepressants, co-segregates with the debrisoquine 4-hydroxylase polymorphism.27 The clinical consequences of genetically determined polymorphic drug metabolism depend on the pharmacologic activity or toxicity of the parent compound compared with that of the metabolites formed by CYP2D6. Clinically important variations can be encountered in the following four situations:
Pharmacologic Effects Are Mediated by the Parent Compound Alone
Mexiletine is a class Ib antiarrhythmic agent that undergoes stereoselective disposition because of an extensive metabolism; less than 10% of an administered oral dose is recovered unchanged in urine.28,29 The major metabolites formed by carbon and nitrogen oxidation are hydroxymethylmexiletine, p-hydroxymexiletine, m-hydroxymexiletine, and N-hydroxymexiletine.28–31 Antiarrhythmic activity resides solely in mexiletine, and all metabolites are inactive. The formation of hydroxymethylmexiletine, p-hydroxymexiletine, and m-hydroxymexiletine is genetically determined and co-segregates with polymorphic debrisoquine 4-hydroxylase (CYP2D6) activity.32 Hence, subjects with the EM phenotype form large amounts of these metabolites. Conversely, clearance of mexiletine is twofold smaller and elimination half-life is longer in subjects with the PM phenotype. Consequently, at the same dose, mean plasma concentrations of mexiletine are higher, and drug accumulation is expected to occur in PM patients during chronic therapy.32 This may lead to side effects such as ataxia and muscle weakness because of the increased block of sodium channels in peripheral nerves.
Combined administration of low-dose quinidine, which is a selective and potent inhibitor of CYP2D6, inhibits mexiletine metabolism through its three CYP2D6 major oxidative pathways and alters mexiletine disposition to such an extent that the pharmacokinetic parameters of the drug are no longer different between EMs and PMs.32 Mexiletine and quinidine have been used in combination to improve antiarrhythmic efficacy and to decrease the incidence of gastrointestinal side effects.33 Because of decreased clearance and increased elimination half-life during quinidine coadministration, EM patients undergoing combined therapy should exhibit higher trough concentrations and lesser peak-to-trough fluctuations in mexiletine plasma concentrations. Drug accumulation and long-term side effects remain a risk if dosage adjustments are not made.
Specific genotypes are associated with metabolic bio-inactivation and, hence, the dose requirement or efficacy of certain drugs. For example, a specific genetic profile (activity of CYP2C9) is associated with higher or lower than average doses required to maintain the INR in the desired range for patients receiving warfarin therapy, and dose prediction based on a pharmacogenetic algorithm is superior to empiric dosing in rapidly achieving the desired target INR.34
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


