CHAPTER 49 Physiology of the Myocardium
THE INTEGRATIVE BIOLOGY OF THE MYOCARDIUM
The essential function of the muscles that make up the myocardium is to transfer to the arteries the volume of blood added to the ventricular chambers during diastole.1 This transfer must occur within the narrow limits of end-diastolic pressures and must produce a flow of materials to the organs that matches the needs of the cells. By needs, we mean matching the flow of oxygen, which is consumed by the cells at a rate greater than all other materials, to the demand for oxygen. By supplying the tissues’ oxygen needs, the demand for all other substances in blood is met. An inevitable consequence of the work done during exercise is an increase in oxygen consumption, and with linear incremental increases in oxygen consumption there are linear incremental increases in cardiac output (CO) to match the increases in venous return (VR). The tight coupling of oxygen demand to CO and VR indicates a regulatory system that is able to sense the tissue oxygen needs, and to engage control mechanisms that adjust the CO. In this chapter, we are concerned with the role of the myocardium in the task of the cardiovascular system to couple oxygen demand to oxygen supply.
In accomplishing this task, the activity of the myocardium must vary over a wide range of short-term regulation (seconds, minutes, and hours) and of long-term regulation (days, weeks, and years). In the short term, during the course of a normal day as tissue oxygen demands and hence cardiac output changes from sleep to strong exercise, variations in activity of the myocardium occur by both intrinsic and extrinsic control mechanisms. The major intrinsic regulator is the Frank-Starling mechanism, in which the pressure developed by the ventricle increases as the end-diastolic volume increases. Extrinsic regulators of the myocardium include the autonomic nerves of the sympathetic and parasympathetic systems, and humoral factors including catecholamines, thyroid hormone, and insulin. In long-term regulation, the activity of the myocardium is more permanently changed in response to chronic changes in oxygen demand associated with frequent bouts of chronic exercise and altered states of the pump and vascular system associated with aging and various long-standing pathologies. In this long-term regulation, the size of the cells making up the ventricular myocardium changes (i.e., hypertrophy or atrophy, without a change in cell number). The cells are remodeled by alterations in subcellular mechanisms regulating contraction and relaxation. This long-term regulation occurs physiologically with normal development of the heart from the immature to the mature myocardium, with normal physiologic aging, and with acquired or inherited pathologies that directly or indirectly affect the function of the myocardium.2,3 In this chapter, we focus on current concepts and theories of the cellular, subcellular, and molecular mechanisms for short-term regulation of the myocardium. These mechanisms are constrained to account for the dynamic and steady-state functional properties of the heart. Changes in flow and ventricular volumes during an episode of exercise reveal these functional properties.
CARDIAC DYNAMICS IN EXERCISE
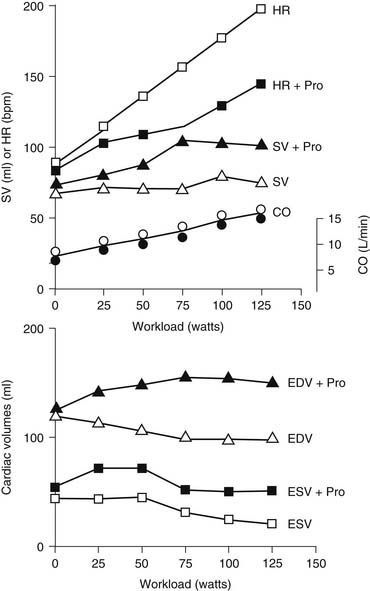
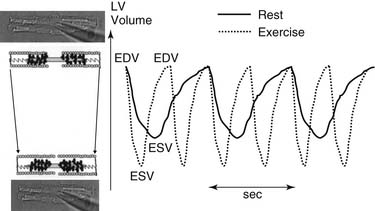
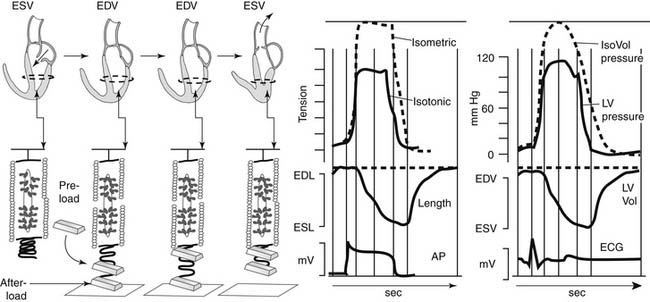
Figure 49-1 displays changes in heart rate (HR), CO, stroke volume (SV), and ventricular volumes of a young healthy adult during a bout of exercise on a stationary bike. The measurements were made before and after administration of propranolol, a β-adrenergic blocking agent. Note that in the control condition, CO increased with workload, but end-diastolic volume (EDV) remained rather constant even though CO nearly tripled. SV increased and end-systolic volume (ESV) decreased. These data show that the increased VR that occurs in exercise is handled by the heart largely by increases in HR and decreases in ESV. Elevations in EDV as a mechanism to increase CO are not favorable because of the increased energy cost according to the law of LaPlace.1 A decrease in ESV provides an important mechanism for matching CO to increased VR without increases in EDV. As we will show, this reduction in ESV at constant EDV can be one measure of the contractile ability of the cells of the heart—that is, the contractility or inotropic state of the heart. After blockade of adrenergic β-receptors with propranolol, there is a blunting of the ability of the sympathetic nervous system to influence the heart. However, CO still increased about threefold. This is testimony to the ability of the cardiovascular system to match the CO to the increased tissue oxygen needs without sympathetic nervous system control mechanisms. However, the increase in CO in the presence of propranolol did not occur without cost. One cost was that the increase in HR was reduced. With a decrease in HR and a constant CO, the SV had to be elevated (CO = SV × HR). The increase in SV occurred largely because of an increase in EDV. Increases in EDV present a threat to the economy of contraction, and they may also stimulate hypertrophic signaling pathways as a result of cell stretch.2,3 These effects of propranolol indicate an important role of the β-receptors and the sympathetic nervous system in regulation of the ability of the heart to maintain CO with little or no change in EDV. Figure 49-2 depicts the dynamics of cardiac function with data relating time dependence of left ventricular volume before and after an episode of exercise. These data demonstrate not only the decrease in ESV with little change in EDV but also enhanced dynamics and abbreviation of the contraction–relaxation cycle. The abbreviation of cardiac cycle time is critical for the maintenance of cardiac filling during the fast HR that occurs with exercise. Figure 49-2 also illustrates that the volume changes are associated with shortening of the cells making up the left ventricular chamber, and that the changes in cell length reflect changes in sarcomere length. We discuss next the molecular and cellular mechanisms responsible for maintenance of CO with elevations of VR with minimal change in EDV.
MOLECULAR CELLULAR BIOLOGY
Figure 49-3 depicts cellular structures involved in excitation, contraction, and relaxation. Tight junctions of low electrical resistance connect heart cells.4,5 When one cell is activated (depolarized), all cells become activated. Therefore, unlike skeletal muscle, the heart does not recruit motor units to regulate contraction. Instead, regulation is at the level of the cells themselves. There are mechanisms that permit regulation of the activity of each cell to meet varying demands on the circulation. We will therefore concentrate now on understanding overt left ventricular cardiac function in terms of the properties of the cardiac myocytes. The objectives are to understand the following:
SARCOMERE MECHANICS
Sarcomeres are the fundamental structural units responsible for the ability of myocardial cells to shorten and generate forces (Fig. 49-4, and see Fig. 49-3). Side arms of the myosin molecules (cross-bridges) that make up the thick filament are molecular motors that also hydrolyze ATP. Light chains on the myosin head, which are different in ventricles and atria, appear to regulate the rate of the ATP hydrolysis. A thick filament–associated protein (called myosin-binding protein C, or C-protein) may be important in regulating the radial movement of the cross-bridge. C-protein also binds to titin, a long structural protein that extends from the center of the sarcomere to the Z-disc. This interaction is of significance in cross-bridge function and in the generation of passive tension. The reaction of the myosin cross-bridges with the actins of the thin filaments generates active cellular force, shortening, and power.6,7 The basic reaction cycle includes an attachment step; a movement of the lever arm of the myosin head, which impels the thin filament in each half-sarcomere to slide toward the center; and a detachment step, which completes the cycle.6 Figure 49-4 displays the cross-bridge in diastole (left) and at the end of the power stroke (right). The energy for these movements comes from the hydrolysis of one molecule of Mg-ATP during each cycle. In diastole, the cross-bridges contain bound Mg-ADP and inorganic phosphate (Pi), which has been generated from the splitting of Mg-ATP on the surface of the myosin head, poising it for reaction with actin. As actin sites become available, the cross-bridge attaches and enters into a catalytic cycle in which the release of Pi and Mg-ADP, together with isomerization of the cross-bridge, induces a progressive change in mechanical state of the cross-bridge, leading to thin-filament sliding. The terminal state is a strongly bound so-called rigor cross-bridge that is free of Pi and nucleotide. Detachment requires binding of Mg-ATP, which is quickly split, without release of products, so that the cycle may begin again if actins remain accessible. Each actin–cross-bridge reaction cycle is therefore powered by the hydrolysis of Mg-ATP.
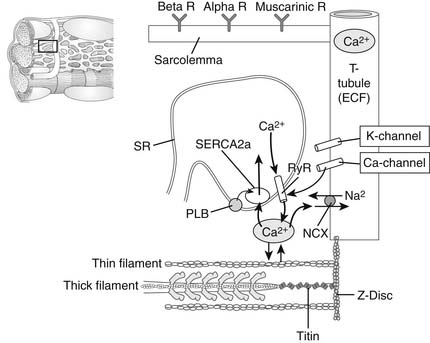
Figure 49–3 Microscopic view of a portion of a myocardial cell illustrating structures critical to excitation–contraction coupling. The T-tubule–containing channels and transporters are shown as an invagination of the surface membrane (or sarcolemma), which contains surface α- and β-receptors for norepinephrine, epinephrine, and muscarinic receptors for acetylcholine. Also shown is the sarcoplasmic reticulum (SR), an internally enclosed network of tubules in which high concentrations of Ca2+ are stored in diastole. With electrical excitation of the cell, Ca2+ channels open and the small release of Ca2+ into the cytoplasm induces Ca2+ release from the SR through ryanodine receptors (RyR2; SR Ca2+ release channels). Ca2+ moves to the myofilaments (shown as a half-sarcomere; see Fig. 49-4) and activates contraction. Ca2+ is removed from the cytoplasm by an SR Ca2+-activated Mg-ATPase (SERCA2a) and exchanged for Na+ by the action of the Na+/Ca2+ exchange protein (NCX) in the sarcolemma. Phospholamban (PLB) inhibits transport of Ca2+ by SERCA2a, and the inhibition is released when PLB becomes phosphorylated. See the text for details. ECF, extracellular fluid.
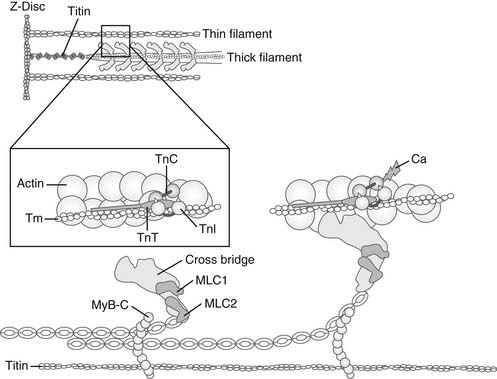
Ca2+ binding triggers conformational changes and movements of the thin-filament proteins troponin (Tn, a heterotrimeric protein complex) and tropomyosin (Tm) that switch on the actin–cross-bridge reaction.3,8 Figures 49-4 and 49-5 illustrate the steps in this process. In diastole, Tn and Tm are situated on the thin filament in positions that hinder the actin–cross-bridge reaction. Tn and Tm are held in this position largely through the tethering action of troponin I (TnI). TnI is an inhibitory protein of the Tn complex, which binds tightly to actin through a highly basic peptide, to the C-terminal end of TnT, the Tm binding unit of Tn, and the C-terminal lobe of troponin C (TnC), the Ca2+ receptor protein. This inhibitory property of TnI is amplified by virtue of these multiple protein–protein interactions to immobilize the long α-helical Tm in a blocking position that encompasses many actins along the thin filament. When released into the myofilament space by mechanisms summarized later, Ca2+ binds to a single regulatory site on the N-terminal lobe of TnC, the Ca2+ receptor protein in the heterotrimeric Tn complex. Ca2+ binding to the C-lobe exposes a “sticky patch” of hydrophobic amino acids that promotes binding of TnC to the inhibitory peptide and C-terminal regions of TnI. This reaction releases the inhibitory peptide from actin and leads to a pivoting of the Tn complex on the thin filament, with TnT acting as a lever to move Tm from its blocking position on the thin filament. An important aspect of the transition from diastole to systole is that the reaction of cross-bridges with the thin filament can itself promote more actin–cross-bridge reactions by cooperative feedback mechanisms. It is apparent that cross-bridge binding may enhance the affinity of TnC for Ca2+ as well as moving Tm farther away from the region of actin that reacts with the cross-bridges.
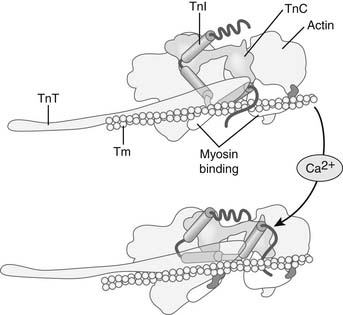
At this stage, it is important to understand that the number of cross-bridges reacting with the thin filaments determines the force generated by the sarcomere. An important determinant of the number of cross-bridges reacting with the thin filaments is the amount of Ca2+ released to the myofilaments and, thus, the relative occupation of sarcomeric TnC proteins with Ca2+ (in the basal state, this is about 20% to 25% of the total TnC).8 Other important determinants of the number of cross-bridges reacting with the thin filaments are the sarcomere length9 and the load (velocity of shortening).6 We will discuss the mechanisms by which each of these variables affects the number of cycling cross-bridges.
Molecular springs interlaced with the thin and thick filaments form elastic elements in the sarcomere that determine passive elastic properties of the cell and have a possible role in active contraction of the cells.10 A major elastic element is the giant protein titin (Fig. 49-3), which is a long and flexible protein extending from the Z-disc to the midline of the sarcomere. As illustrated in Figure 49-3, titin has a region near the Z-disc that is coiled much like a spring. There is accumulating and solid evidence that when the sarcomere is stretched, titin elongates, giving rise to passive tension. Moreover, there is evidence that when the sarcomere shortens, the titin spring imposes a restoring force that is likely to be important in early diastole. Regions of titin in the thin filament–thick filament overlap zone also interact with myosin-binding protein C, a thick filament–associated protein that binds to the head/neck region of myosin. Thus, the conformational changes in titin may also affect cross-bridge disposition. Although not depicted in Figure 48-3, the Z-disc of the sarcomere not only anchors the thin filaments but also links sarcomeres in series by interactions between titin and the thin filaments. There are also lateral connections linking the sarcomere to the surface membrane. In addition to its role in force transmission, the Z-disc is emerging as a locus of communication in the cells. The Z-disc appears as a crossroad for interactions among many diverse proteins including channels, kinases, and phosphatases, and cytoskeletal elements that connect to the nucleus as well as to a network of cytoskeletal proteins and membrane proteins at focal adhesion complexes.10
We will use Figure 49-6, which relates the circumferential shortening associated with the heartbeat to sarcomeric activity, to discuss the cellular and sarcomeric correlates of preload, afterload, and contractility. Thus, in Figure 49-6, we relate the contraction–relaxation cycle of the heart beat to events in a single sarcomere; mechanical changes that occur in the active and passive elements of the sarcomere are related to the beat of the heart. The sarcomere is depicted as a contractile element in series with a passive spring that is a lumped elastic element (collagen, titin, and cytoskeletal proteins). A load attached to the end of the sarcomere establishes the sarcomere length before activation and is termed the preload. The preload stretches the sarcomere to its diastolic length; the correlate of the preload in the ventricle is the EDV. In the ventricle, an end-diastolic pressure (EDP) develops as the passive springs are stretched.
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


