Pharmacologic Therapy for Acute Coronary Syndromes: Introduction
Acute coronary syndromes (ACS), including unstable angina (UA), non–ST-segment elevation myocardial infarction (NSTEMI), and ST-segment elevation myocardial infarction (STEMI), represent the clinical complications of atherosclerosis mediated by plaque rupture1 and superficial endothelial cell erosion2 that produce nonocclusive and occlusive thrombus formation. NSTEMI accounts for more than 1.6 million annual admissions, representing up to 75% of all cases of myocardial infarction (MI) in US hospitals.3 Appropriate care for patients with ACS is informed by a wealth of recent randomized controlled trials, the findings of which have been summarized into national clinical practice guidelines by the American College of Cardiology/American Heart Association (ACC/AHA),3-5 the American College of Chest Physicians (ACCP),6-8 and the European Society of Cardiology (ESC).9,10 This chapter will review the pharmacology of antiplatelet, anticoagulant, and fibrinolytic agents and also focus on evidence-based recommendations for antithrombotic therapy for ACS. Since the prior edition, assessing the net clinical benefit of a particular antithrombotic agent—namely, balancing efficacy and safety (ie, bleeding)—has moved front and center in our analysis of clinical trials.11-13 Rather than focusing solely on ischemic events (eg, recurrent MI, stroke), attention has shifted to the importance of bleeding, given its high prevalence and adverse impact on mortality, which is likely secondary to multiple factors including impaired oxygen delivery, adverse effects of transfusion (ie, proinflammatory, old vs new blood), and cessation of antithrombotic medications.11,12,14 There are important sex- and age-related factors that promote bleeding due to excessive dosing.15,16 Understanding these pharmacologic considerations is essential for the practicing clinician. Finally, guideline adherence rate is significantly associated with in-hospital mortality. In an observational analysis of hospital care in 350 academic and nonacademic US centers of 64,775 patients enrolled in the CRUSADE (Can Rapid Risk Stratification of Unstable Angina Patients Suppress Adverse Outcomes With Early Implementation of the ACC/AHA Guidelines) trial, every 10% increase in composite adherence at a hospital was associated with an analogous 10% decrease in its patients’ likelihood of in-hospital mortality.17 Thus a thorough knowledge of pharmacologic therapy and guideline-recommended treatments is the foundation for optimizing outcomes for patients with ACS.
Antiplatelet Therapy
Currently, there are three different classes of antiplatelet agents that are approved for the treatment and/or prevention of recurrent events in patients with ACS: cyclooxygenase-1 (COX-1) inhibitors (aspirin), adenosine diphosphate (ADP) P2Y12 receptor antagonists (thienopyridines and nonthienopyridines), and glycoprotein (GP) IIb/IIIa inhibitors. These agents are mostly used in combination in patients presenting with an ACS, in particular those undergoing percutaneous coronary intervention (PCI). Details of each of these classes of antiplatelet agents and their indications for use are provided below. In addition, limitations of the most frequently used oral antiplatelet agents (antiplatelet drug “resistance”), as well as novel antiplatelet therapies under advanced clinical investigation, which may represent treatment alternatives to current strategies, are described.
Aspirin (acetylsalicylic acid [ASA]) is one the most important medications used in cardiovascular medicine. In particular, aspirin irreversibly inactivates cyclooxygenase (COX) activity of prostaglandin H (PGH) synthase 1 and synthase 2, also referred to as COX-1 and COX-2, respectively.18,19 These isozymes catalyze the conversion of arachidonic acid to PGH2, an unstable intermediate that is the substrate for several downstream isomerases that lead to the generation of several prostanoids, including thromboxane A2 (TXA2) and prostacyclin (PGI2) (Fig. 61–1A).18,19 Aspirin diffuses through the cell membrane and enters the COX “channel,” which is a narrow hydrophobic channel connecting the cell membrane to the catalytic pocket of the COX enzyme. Aspirin acetylates a serine residue (serine 529 in human COX-1 and serine 516 in human COX-2) located in the narrowest section of the channel impeding arachidonic acid from gaining access to the catalytic site of the COX enzyme. Higher doses of aspirin are needed to inhibit COX-2 than to inhibit COX-1. These differences explain why very high doses of aspirin are needed to achieve anti-inflammatory and analgesic effects, whereas low doses of aspirin lead to antiplatelet effects. Vascular endothelial cells and newly formed platelets express both COX-1 and COX-2. However, only COX-1 is expressed in mature platelets. Importantly, TXA2 (an amplifier of platelet activation and a vasoconstrictor) is derived largely from COX-1 (mostly from platelets), and its biosynthesis is highly sensitive to inhibition by aspirin, whereas vascular PGI2 (a platelet inhibitor and a vasodilator) is derived predominantly from COX-2 and is less susceptible to inhibition by low doses of aspirin. Therefore, low-dose aspirin ultimately preferentially blocks platelet formation of TXA2, diminishing platelet aggregation mediated by thromboxane (TP) receptor pathways. Aspirin may also inhibit thrombosis via acetylation of guanosine triphosphate binding proteins, thrombin receptors, and prothrombin.20
Figure 61–1.
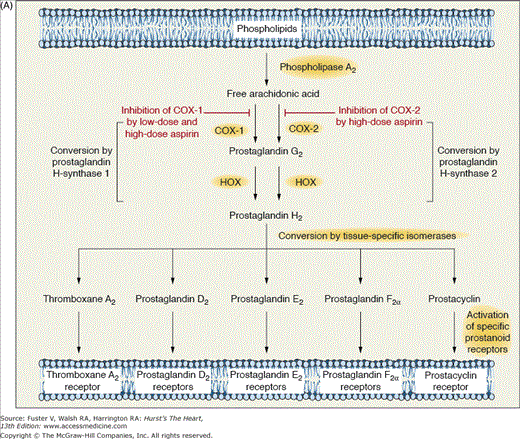
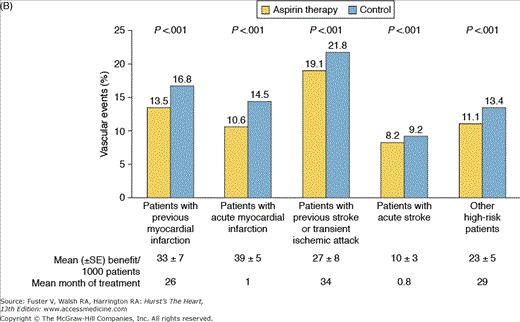
A. Mechanism of action of aspirin. Arachidonic acid, a 20-carbon fatty acid containing four double bonds, is released from membrane phospholipids by several forms of phospholipase A2, which are activated by diverse stimuli. Arachidonic acid is converted by cytosolic prostaglandin H synthases, which have both cyclooxygenase and hydroperoxidase (HOX) activity, to the unstable intermediates prostaglandin G2 and prostaglandin H2, respectively. The synthases are also termed cyclooxygenases and exist in two forms, cyclooxygenase-1 (COX-1) and cyclooxygenase-2 (COX-2). Low-dose aspirin selectively inhibits COX-1, whereas high-dose aspirin inhibits both COX-1 and COX-2. Prostaglandin H2 is converted by tissue-specific isomerases to multiple prostanoids. These bioactive lipids activate specific cell-membrane receptors of the superfamily of G-protein–coupled receptors, such as the thromboxane receptor, the prostaglandin D2 receptors, the prostaglandin E2 receptors, the prostaglandin F2a receptors, and the prostacyclin receptor. B. Absolute effects of antiplatelet therapy with aspirin on the risk of vascular events. The benefit of aspirin therapy in reducing vascular events, including nonfatal myocardial infarction, nonfatal stroke, or death from vascular causes, in five groups of high-risk patients is shown. The figure is based on an analysis of data from the Antithrombotic Trialists’ Collaboration. Reproduced with permission from Patrono et al.19
Aspirin is rapidly absorbed in the upper gastrointestinal tract and is associated with measurable platelet inhibition within 60 minutes.18,19 The plasma half-life of aspirin is approximately 20 minutes, and peak plasma levels occur 30 to 40 minutes after ingestion. Enteric-coated aspirin delays absorption, with peak plasma levels at 3 to 4 hours.21 Because the blockade of COX-1 induced by aspirin is irreversible, COX-mediated TXA2 synthesis is prevented for the entire life span of the platelet (~7-10 days). Therefore, even low doses of aspirin can produce long-lasting platelet inhibition.
The optimal dose of aspirin for prevention of cardiovascular events has been subject of controversy, and it is not firmly defined. Pharmacodynamic studies show that aspirin may be effective in inhibiting the COX-1 enzyme at doses as low as 30 mg/d.18,19 The Antiplatelet Trialists’ Collaboration showed that a daily oral aspirin dose of 75 to 150 mg is as effective as higher doses for long-term treatments, whereas aspirin doses <75 mg have been less widely assessed in clinical trials, and their use is generally not recommended.22,23 Importantly, higher doses of aspirin (>150 mg) do not offer greater protection from recurrent ischemic events.22,23 When given in combination with clopidogrel, which is the standard of care in patients with ACS, the dose of aspirin should generally be lowered to 75 to 100 mg. This is based on a post-hoc analysis of data from the CURE (Clopidogrel in Unstable Angina to Prevent Recurrent Events) study in which similar efficacy but less major bleeding was seen in the low-dose (<100 mg) aspirin group.24 Similarly, low-dose (<100 mg) aspirin should be considered in patients requiring concomitant treatment with an anticoagulant (eg, vitamin K antagonists). More research is needed to better define the optimal dose of aspirin and the impact on clinical events, especially when it is combined with other antithrombotic agents.
Overall, the results of biochemical studies on its mechanism of action, the lack of dose-response relationship in clinical studies evaluating its antithrombotic effects, and the dose dependence of its side effects all support the use of low-dose aspirin. Despite this evidence, practice patterns in some countries still largely use high-dose aspirin (eg, 300-325 mg) based on guideline recommendations for drug-eluting stents. The first large-scale prospective randomized study to compare high- versus low-dose aspirin was the CURRENT/OASIS-7 (Clopidogrel Optimal Loading Dose Usage to Reduce Recurrent Events—Organization to Assess Strategies in Ischemic Syndromes) trial,25 which included patients with ACS (n = 25,087) scheduled to undergo angiography. The study had a 2 × 2 factorial design, and patients were randomized in a double-blind fashion to high (600-mg loading dose followed by 150 mg daily for 7 days and then 75 mg daily thereafter) or standard (300-mg loading dose followed by 75 mg daily thereafter) doses of clopidogrel for a month and also included an open-label randomization to high-dose (300-325 mg daily) versus low-dose (75-100 mg daily) of aspirin. The trial did not show significant differences in efficacy between high- and low-dose aspirin. Although there were no differences in major bleeds between the two aspirin doses, a trend toward a higher rate of gastrointestinal bleeds in the high-dose group (0.38% vs 0.24%; P = .051) was observed. It should be taken into consideration that this trial lasted only 1 month, whereas studies showing increased bleeding with high-dose aspirin were typically of longer duration.
Clinical trials and expert consensus statements evaluating the use of aspirin for primary prevention of cardiovascular events are controversial and beyond the scope of this chapter.26-32 On the contrary, aspirin is still the antiplatelet drug of choice for secondary prevention of recurrent ischemic events in patients with various clinical manifestations of coronary artery disease (CAD), including stable CAD and ACS, and those undergoing coronary revascularization (percutaneous or surgical).3-5 The benefit of aspirin therapy in reducing vascular events including nonfatal MI, nonfatal stroke, or death from vascular causes in five groups of high-risk patients is illustrated in Fig. 61–1B. In these patients, particularly those with ACS or undergoing PCI, aspirin should be given as promptly as possible, at an initial dose of 162 to 325 mg, followed by a daily dose of 75 to 162 mg.3-5
The benefit of aspirin therapy in the management of patients with ACS has been repeatedly demonstrated in earlier trials. There are four randomized trials demonstrating beneficial effects of aspirin in patients with UA/NSTEMI showing an approximately 50% reduction in the risk of death or MI.33-36 The Swedish angina pectoris aspirin trial, in which patients (n = 2035) were allocated to receive 75 mg of aspirin daily or placebo,37 showed that aspirin led to significant reductions in death and MI among patients with UA (46% reduction), those undergoing PCI (53% reduction), and those with stable angina (33% reduction). In the setting of STEMI, aspirin was found to decrease the rate of angiographic reocclusion by more than 50% in a meta-analysis of 32 angiographic trials.38 In the Second International Study of Infarct Survival (ISIS-2)39 trial, STEMI patients (n = 17187) presenting within 24 hours of the onset of symptoms were randomized to receive intravenous (IV) streptokinase, 162.5 mg of aspirin daily for 30 days, both, or neither. In patients receiving aspirin therapy alone, there was a significant 23% reduction in vascular mortality and a nearly 50% reduction in the risk of nonfatal re-infarction and nonfatal stroke at the end of 5 weeks. This benefit occurred irrespective of heparin use.
The benefits of aspirin in reducing recurrent ischemic events, including cardiovascular death, MI, and stroke in patients with CAD,22,23 has also led to the near-universal use of this medication for patients undergoing PCI, which currently represents the predominant management strategy in patients with ACS (60%-70% of ACS patients undergo PCI). The initial studies involving aspirin in PCI included combined antiplatelet regimens with dipyridamole or ticlopidine. The combination of aspirin plus dipyridamole was shown to reduce the incidence of periprocedural MI during PCI by 77% compared with patients receiving placebo when administered 24 hours before PCI and continued for 4 to 7 months.40 Dipyridamole was shown to provide no additional benefit beyond that conveyed by aspirin alone during elective angioplasty.41 Aspirin has been shown to be effective in reducing recurrent ischemic events in patients undergoing intracoronary stent placement, especially in combination with a thienopyridine.42-45
Aspirin is recommended for patients who undergo coronary artery bypass grafting (CABG). CABG with saphenous vein grafts is associated with a 5% to 15% graft occlusion rate during the first postoperative month.46,47 In the immediate postoperative period, aspirin reduces the rate of early thrombotic graft occlusion by approximately 50%, and continued aspirin therapy for 1 year further decreases occlusive events.46 Although there is no evidence of the additional benefit of aspirin on long-term graft patency after 1 year of therapy,48 prolonged aspirin therapy is required for secondary prevention of atherothrombotic events in this group of patients with proven, complex coronary artery disease.
In the Antithrombotic Trialists’ Collaboration meta-analysis,22,23 the proportional increase in risk of a major extracranial bleed with antiplatelet therapy was approximately 60% (odds ratio 1.6; 1.4-1.8, 95% CI). The proportional increase in fatal bleeds was not significantly different from that for nonfatal bleeds; however, only the excess of nonfatal bleeds was significant.49 In one small study, aspirin (75-325 mg/d) was associated with significant decrease in creatinine clearance and decrease in uric acid excretion after 2 weeks of therapy in elderly patients.50 Three types of aspirin sensitivity have been described: respiratory sensitivity (asthma and/or rhinitis), cutaneous sensitivity (urticaria and/or angioedema), and systemic sensitivity (anaphylactoid reaction).22,23 The prevalence of aspirin-exacerbated respiratory tract disease is approximately 10%, and for aspirin-induced urticaria, the prevalence varies from 0.07% to 0.2% in the general population.51 In patients with CAD presenting with allergy or intolerance to aspirin, clopidogrel is the treatment of choice.3 Desensitization using escalating doses of oral aspirin can also be performed and may be an option in patients requiring long-term dual antiplatelet therapy with aspirin and clopidogrel, particularly those treated with drug-eluting stents (DES).52 This approach should be confined to patients for whom adherence to continued aspirin has a very high likelihood due to the risks associated with discontinuation followed by reintroduction of aspirin after desensitization.
Platelet ADP signaling pathways mediated by the P2Y1 and P2Y12 receptors play a central role in platelet activation and aggregation.53-55 The thienopyridine derivatives selectively and irreversibly inhibit the P2Y12 ADP receptor (Fig. 61–2).53-55 Although both receptors are needed for aggregation, ADP-stimulated effects on platelets are mediated mainly by Gi-coupled P2Y12 receptor activation, which leads to sustained platelet aggregation and stabilization of the platelet aggregate, whereas P2Y1 is responsible for an initial weak and transient phase of platelet aggregation and change in platelet shape.55,56 In particular, P2Y1 is coupled to a Gq protein, which regulates activation of phospholipase C, resulting in diacylglycerol and inositol triphosphate production. Diacylglycerol activates protein kinase C, leading to phosphorylation of myosin light chain kinase and granule secretion; inositol triphosphate leads to mobilization of intracellular calcium. The P2Y1 receptor is coupled to another G protein that leads to change in platelet shape. Activation of the P2Y12 receptor leads to a complex series of intracellular signaling events that culminate in activation of the platelet GP IIb/IIIa receptor, granule release, amplification of platelet aggregation, and stabilization of the platelet aggregate. The P2Y12 receptor is coupled to a Gi protein, which regulates activation of phosphoinositide-3-kinase and inhibition of adenylyl cyclase. Phosphoinositide-3-kinase activation leads to GP IIb/IIIa activation through activation of intraplatelet kinases, whereas inhibition of adenylyl cyclase decreases cyclic adenosine monophosphate (cAMP) levels. Reduction of cAMP levels modulates the activity of cAMP-dependant protein kinases, which in turn reduce cAMP-mediated phosphorylation of vasodilator-stimulated phosphoprotein and eliminate its protective effect on GP IIb/IIIa receptor activation.55,56
Figure 61–2.
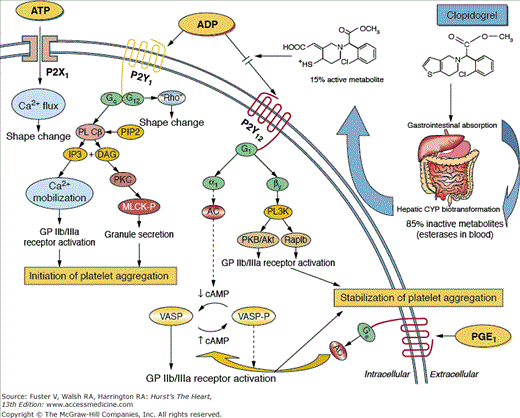
P2 receptors and mechanism of action of clopidogrel. Clopidogrel is a prodrug administered orally. Approximately 85% of the prodrug is hydrolyzed by esterases in the blood to an inactive carboxylic acid derivative and only 15% of the prodrug is metabolized by the cytochrome P450 (CYP) system in the liver to generate an active metabolite. The active metabolite irreversibly inhibits the adenosine diphosphate (ADP) P2Y12 receptor. Activation of the P2X1 and P2Y1 receptors lead to alteration in shape and initiate a weak and transient phase of platelet aggregation. The P2X1 mediates extracellular calcium influx and utilizes adenosine triphosphate (ATP) as an agonist. The binding of ADP to the Gq-coupled P2Y1 receptor leads to activation of phospholipase C (PLC), which generates diacylglycerol (DAG) and inositol triphosphate (IP3) from phosphatidylinositol bisphosphate (PIP2). DAG activates protein kinase C (PKC), leading to phosphorylation of myosin light chain kinase (MLCK-P); IP3 leads to mobilization of intracellular calcium. The P2Y1 receptor is coupled to another G-protein which can lead to change in platelet shape. The binding of ADP to the Gi-coupled P2Y12 receptor liberates the Gi protein subunits αi and β and results in stabilization of platelet aggregation. The αi subunit leads to inhibition of adenylyl cyclase (AC), which reduces cyclic adenosine monophosphate (cAMP) levels. This in turn diminishes cAMP-mediated phosphorylation of vasodilator-stimulated phosphoprotein (VASP) (VASP-P). The status of VASP-P modulates glycoprotein (GP) IIb/IIIa receptor activation. The subunit βγ activates the phosphatidylinositol 3-kinase (PI3K), which leads to GP IIb/IIIa receptor activation through activation of kinases. Prostaglandin E1 (PGE1) activates AC, which increases cAMP levels and status of VASP-P. Solid arrows indicate activation. Dotted arrows indicate inhibition. Reproduced with permission from Angiolillo et al.222
Several families of P2Y12 inhibitors have been developed for clinical use. However, only thienopyridines (ticlopidine, clopidogrel, and prasugrel), which are nondirect, orally administered, and irreversible P2Y12 receptor inhibitors, are currently approved for clinical use. When given in combination with ASA, thienopyridines have a synergistic effect and therefore achieve greater platelet inhibition than either agent alone.57,58 The inhibition of platelet aggregation by thienopyridines is concentration-dependent. However, thienopyridines are prodrugs and are thus inactive in vitro and need to be metabolized by the hepatic cytochrome P450 (CYP) system in order to produce an active metabolite capable of selectively inhibiting the P2Y12 receptor. Because blockade of P2Y12 is irreversible, platelet inhibitory effects induced by thienopyridines last for the entire life span of the platelet.
Ticlopidine was the first thienopyridine to be developed and was approved for clinical use in 1991. Ticlopidine achieves significant platelet inhibition after 2 to 3 days of therapy at the approved dose of 250 mg twice daily. Ticlopidine showed its superiority in combination with aspirin when compared with aspirin alone or anticoagulation in combination with aspirin in a number of trials for prevention of recurrent ischemic events in patients undergoing PCI.42-45 However, due to safety concerns (mainly high rates of neutropenia) and twice-daily dosing, ticlopidine has been largely replaced by clopidogrel (a second-generation thienopyridine) because of its better safety profile.59 Pooled data from more than 10,000 patients undergoing PCI demonstrated that clopidogrel was associated with a significant reduction in the incidence of major adverse cardiac events, including mortality, compared with ticlopidine.60 Only one randomized trial demonstrated superiority of ticlopidine over clopidogrel in stented patients with regard to the combined end point of death or MI.61
Clopidogrel, a second-generation thienopyridine, differs structurally from ticlopidine by the addition of a carboxymethyl group.53-56 The inhibition of platelet aggregation by clopidogrel is also concentration-dependent. Clopidogrel is an inactive prodrug that requires oxidation by the CYP system to generate an active metabolite. However, approximately 85% of the prodrug is hydrolyzed by esterases to an inactive carboxylic acid derivative. Therefore, only 15% of the prodrug is metabolized by the CYP system into an active metabolite. In particular, the thiophene ring of clopidogrel is oxidized to form an intermediate metabolite (2-oxo-clopidogrel), which is further oxidized, resulting in the opening of the thiophene ring and the formation of a carboxyl and thiol group. CYP3A4, CYP3A5, CYP2C9, and CYP1A2 are involved in one oxidation step; CYP2B6 and CYP2C19 are involved in both steps. The reactive thiol group of the active metabolite of clopidogrel forms a disulfide bridge between one or more cysteine residues of the P2Y12 receptor, resulting in its irreversible blockade. Although clopidogrel has a half-life of only 8 hours, it has an irreversible effect on platelets that lasts from 7 to 10 days.
Compared with ticlopidine, clopidogrel has the advantage of being able to be administered as a loading dose, allowing antiplatelet effects to be achieved within hours after administration. The approved loading and maintenance doses of clopidogrel are 300 mg and 75 mg, respectively. However, numerous pharmacodynamic studies have shown that higher doses can achieve greater platelet inhibition. In particular, most of these studies have been performed in patients undergoing PCI and evaluated high loading dose regimens. Most studies have consistently shown that high (≥600 mg) loading dose regimens are associated with faster and more potent platelet inhibitory effects as compared with 300 mg.62-65 A high loading dose regimen has also been associated with a reduction in periprocedural MI in patients undergoing PCI.25,66-68
The recommended loading dose of clopidogrel is 300 to 600 mg in the setting of PCI, including patients undergoing primary PCI in the context of STEMI.3-5 A 300-mg loading dose of clopidogrel should also be given in patients with STEMI treated with fibrinolytic therapy. This should be followed by a maintenance dose of 75 mg daily. No dosage adjustment is necessary for patients with renal impairment, including patients with end-stage renal disease. These recommendations have been done in light of the results of several large-scale clinical trials that have shown a clear benefit of clopidogrel in addition to aspirin in terms of preventing recurrent ischemic events, including stent thrombosis, when compared with aspirin alone.69-74 In patients undergoing PCI for ACS, clopidogrel 75 mg daily should be given for at least 12 months.3-5 This recommendation is independent of revascularization strategy (ie, similar for bare-metal stent [BMS], DES, or balloon angioplasty). If the risk of morbidity because of bleeding outweighs the anticipated benefit afforded by thienopyridine therapy, earlier discontinuation should be considered. In patients undergoing DES placement, continuation of clopidogrel beyond 1 year may also be considered on an individualized basis. In patients taking a thienopyridine in whom CABG is planned and can be delayed, it is recommended that the drug be discontinued to allow for dissipation of the antiplatelet effect. The period of withdrawal should be at least 5 days in patients receiving clopidogrel.3-5
In patients undergoing PCI, clopidogrel loading dose should be given as early as possible.3-5 Pretreatment with clopidogrel before PCI improves 30-day outcomes compared with those not pretreated.70,71 In a meta-analysis of the results of three randomized trials, clopidogrel pretreatment before PCI is beneficial and safe, regardless of whether a GP IIb/IIIa inhibitor is used at the time of PCI.75 In order to achieve the pretreatment benefit of early clopidogrel, patients must be treated at least >6, and preferably >12, hours before PCI.76 Use of a 600-mg clopidogrel load may allow one to reduce the pretreatment period to as short as 2 hours before PCI.77
The Clopidogrel versus Aspirin in Patients at Risk of Ischemic Events (CAPRIE) trial evaluated the efficacy of clopidogrel (75 mg daily) versus aspirin (325 mg daily) in reducing the risk of ischemic outcomes in patients (n = 19,185) with a history of recent MI, recent ischemic stroke, or established peripheral artery disease. The overall results showed a significantly lower annual rate of the composite end point (vascular death, MI, or ischemic stroke) with clopidogrel (5.32% vs 5.83%; P = .043).78 Because aspirin and clopidogrel act through different mechanisms to inhibit platelet activity, the potential synergy of these two agents has been explored in several large-scale clinical trials, particularly in high-risk patients, showing greater efficacy of this strategy.69-74
The pivotal ACS and PCI trials comparing dual antiplatelet therapy with aspirin and clopidogrel versus aspirin alone are summarized in Table 61–1. The CURE trial evaluated the short-term and long-term effects of clopidogrel in combination with aspirin compared with aspirin alone in the prevention of ischemic complications in patients (n = 12,562) with UA/NSTEMI.69 Clopidogrel plus aspirin provided a 20% relative risk reduction in the primary composite outcome (cardiovascular death, nonfatal MI, or stroke) compared with aspirin alone. The long-term clinical benefit associated with dual antiplatelet therapy with aspirin and clopidogrel was independent of coronary revascularization and present in both medically managed patients and those undergoing PCI or CABG. More patients in the clopidogrel plus aspirin group than in the aspirin alone group showed major bleeding (3.7% vs 2.7%; relative risk, 1.38; P = .001). Although all groups showed an advantage with clopidogrel plus aspirin, the benefit appeared greatest in the high-risk group.79 A subgroup analysis of patients (n = 2658) from the CURE study who underwent PCI (PCI-CURE) showed that clopidogrel pretreatment significantly reduced post-PCI cardiovascular death or MI, with a 31% overall relative risk reduction (P = .03).70 The Clopidogrel for the Reduction of Events During Observation (CREDO) trial evaluated the benefit of clopidogrel in patients undergoing elective PCI.71 Patients who were given aspirin plus clopidogrel for 12 months experienced a 26.9% (P = .02) reduction in the relative risk of vascular events versus aspirin alone. A prespecified subgroup analysis of patients pretreated with a 300-mg clopidogrel loading dose at least 6 hours before PCI suggested a 38.6% (P = .051) reduction in periprocedural adverse events. The CURE, PCI-CURE, and CREDO trials all reported long-term benefit of dual antiplatelet therapy (9-12 months) over single anti-platelet therapy in patients with ACS. The benefit of clopidogrel has also been demonstrated in patients with STEMI in the Clopidogrel and Metoprolol in Myocardial Infarction (COMMIT) and Clopidogrel as Adjunctive Reperfusion Therapy (CLARITY) trials, when given in addition to aspirin or aspirin plus fibrinolytic therapy (see Table 61–1).72,73
| Trial Name | n | Patients | Treatment (mg/d) | Primary End Point | Event Rate (Active Treatment vs Control [%]) | P |
|---|---|---|---|---|---|---|
| CAPRIE | 19,185 | Previous stroke or MI or symptomatic PAD | Clopidogrel vs ASA | Ischaemic stroke, MI, or vascular death at 1 y | 5.3% vs 5.8% | 0.043 |
| CURE | 12,562 | NSTE ACS, unstable angina | Clopidogrel + ASA vs ASA | CV death, nonfatal MI, and stroke at 1 y | 9.3% vs 11.4% | <0.001 |
| CREDO | 2116 | ACS with PCI | Clopidogrel + ASA vs ASA | CV death, MI, or stroke at 1 y | 8.5% vs 11.5% | 0.02 |
| PCI-CURE | 2658 | NSTE ACS with PCI | Clopidogrel + ASA vs ASA | CV death, MI, or revascularization within 30 d | 4.5% vs 6.4% | 0.03 |
| CLARITY | 3491 | STEMI | Clopidogrel + ASA + FA vs ASA + FA | Occluded infarct-related artery, death or recurrent MI before angiography | 15.0% vs 21.7% | <0.001 |
| COMMIT | 45,852 | STEMI | Clopidogrel + ASA vs ASA | CV death, reinfarction, or stroke at 28 d | 9.2% vs 10.1% | 0.002 |
| CHARISMA | 15,603 | CVD or multiple risk factors | Clopidogrel + ASA vs ASA | MI, stroke, or CV death at 28 mo | 6.8% vs 7.3% | 0.22 |
| CURRENT-OASIS 7 | 25, 087 | ACS with planned early invasive management with intended PCI | Double-dose clopidogrel + ASA vs low-dose clopidogrel + ASA | CV death, MI, or stroke at 30 d | 4.2% vs 4.4% (overall cohort) | 0.37 |
In contrast to the positive results of the trials described above, the Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management, and Avoidance (CHARISMA) trial showed that after 28 months, addition of clopidogrel to aspirin was not better than aspirin alone in reducing the primary composite end point (cardiovascular death, MI, or stroke) in patients (n = 15,603) with cardiovascular disease or multiple cardiovascular risk factors (see Table 61–1).80 Increased bleeding and higher mortality rates were reported when patients with multiple risk factors alone (n = 3284) were analyzed, providing evidence that dual antiplatelet therapy should not be used in patients without a history of established vascular disease. In the subgroup of patients with clinically evident atherothrombosis (n = 12,153), there was a 12% relative risk reduction in event rates with clopidogrel (P = .046). A subsequent analysis of CHARISMA data from patients (n = 9478) with prior MI, stroke, or symptomatic peripheral arterial disease, also known as “CAPRIE-like,” showed that a dual antiplatelet regimen offered significant benefit, reducing the relative rate for ischemic events by 17% (P = .01).81 Taken together, the results of the clinical trial experience with clopidogrel suggest that dual antiplatelet therapy is useful in high-risk settings, such as in patients with various clinical manifestations of ACS or undergoing PCI, and has not been proven beneficial in lower risk patients.
Most recently, the CURRENT/OASIS 7 trial evaluated the efficacy and safety of double-dose clopidogrel in patients (n = 25,087) with ACS.25 In this trial, double-dose clopidogrel was defined as a 600-mg loading dose and 150 mg once daily for 7 days, followed by 75 mg once daily. Standard-dose clopidogrel was defined as a 300-mg loading dose, followed by 75 mg once daily. Patients were also randomized to receive low-dose (75-100 mg/d) or high-dose (300-325 mg/d) aspirin. In the overall study population, there was no significant difference in the primary end point (composite of cardiovascular death, MI, or stroke) at 30 days between patients receiving double-dose versus standard-dose clopidogrel (4.4% vs 4.2%; P = .37). However, two positive interactions were encountered, which included PCI and high-dose aspirin. In patients who underwent PCI (n = 17,232), double-dose clopidogrel was associated with a significant 15% relative reduction (P = .036) in the primary end point and a 42% relative reduction (P = .001) in definite stent thrombosis versus the standard-dose regimen. There was a modest excess in CURRENT-defined major bleeds, but no difference in Thrombolysis in Myocardial Infarction (TIMI) major bleeds, intracranial hemorrhage (ICH), fatal bleeds, or CABG-related bleeds between the two clopidogrel treatment arms. In addition, there was an interaction with aspirin dose, whereas patients receiving high-dose clopidogrel had better outcomes when treated with high-dose aspirin. The explanation to the latter finding remains unknown.
An intense area of investigation with DES has been whether the risk of late (30 days to 1 year) and very late (beyond 1 year) stent thrombosis may be diminished by the extended use of dual antiplatelet therapy with aspirin and clopidogrel. In fact, analysis of key randomized trials comparing DES with BMS through 5-year follow-up have demonstrated a numerically higher incidence of very late stent thrombosis with DES compared with BMS,82-84 and observational studies have indicated a higher risk of stent thrombosis in off-label indications for BMS and DES.85,86 Because of the important prognostic implications associated with stent thrombosis with fatality rates in up to 40% to 50% of patients,87 as a precautionary measure and based on trials associating thienopyridine discontinuation within the first 6 months with risk of DES thrombosis, guideline recommendation advocated 12 months of dual antiplatelet therapy after DES placement in patients without contraindications and bleeding risk.4,5 However, this recommendation was not based on any prospective randomized trial evidence associating extended duration of dual antiplatelet therapy with a reduction in late stent thrombosis, and there is evidence from registry data that prolonging dual antiplatelet therapy beyond 6 months is not clearly associated with a reduction in stent thrombosis.88-92 Nevertheless, based on data from randomized trials evaluating clopidogrel in PCI trials with BMS70,71 and from observational studies indicating lower risk of death and MI with long-term dual antiplatelet therapy,93-96 the safety concerns have led expert consensus statements to recommend at least 12 month of treatment.4,5
There are several ongoing trials aiming to investigate the optimal duration of dual antiplatelet therapy in DES-treated patients. The DAPT (Dual Anti Platelet Study, NCT00977938) is currently randomizing 20,645 patients free from events at 12 months to placebo versus an additional 18 months of thienopyridine treatment (clopidogrel or prasugrel). The primary end points will address the 33-month incidence of death, MI, or stroke and the incidence of definite or probable stent thrombosis, providing strong evidence to support, or discourage, extended use of thienopyridines beyond 12 months. The ISAR-SAFE (Safety and Efficacy of Six-Month Dual Antiplatelet Therapy After Drug-Eluting Stenting, NCT00661206) will compare the efficacy of the 6- and 12-month strategies in reducing the rate of death, MI, stroke, and major bleeding at 15 months in 6,000 6-month event-free patients. Further insights on the relative efficacy of a 12- versus 24-month approach will be explored by the REAL-LATE (Correlation of Clopidogrel Therapy Discontinuation in REAL-World Patients Treated With Drug-Eluting Stent Implantation and Late Coronary Arterial Thrombotic Events, NCT00484926) and the ZEST-LATE (Evaluation of the Long-Term Safety After Zotarolimus-Eluting Stent, Sirolimus-Eluting Stent, or Paclitaxel-Eluting Stent Implantation for Coronary Lesions—Late Coronary Arterial Thrombotic Events, NCT00590174) trials. These trials will shed more light on the efficacy (ie, reduction of ischemic events, including stent thrombosis) as well as the safety (ie, bleeding) of prolonging dual antiplatelet therapy beyond 12 months.
In the clopidogrel aspirin stent international cooperative study (CLASSICS), major peripheral or bleeding complications were similar between clopidogrel (1.3%) and ticlopidine (1.2%).59 In CAPRIE, gastrointestinal hemorrhage occurred at a rate of 2.0% and 2.7% in patients treated with clopidogrel and aspirin, respectively.78 The incidence of ICH was 0.4% for clopidogrel compared with 0.5% for aspirin. In CURE, clopidogrel use with aspirin was associated with an increase in bleeding compared with placebo with aspirin (3.7% vs 2.7%; relative risk, 1.38; P = .001).69 The risk of bleeding, however, was increased in patients using higher doses of aspirin. There was an excess in major bleeding in patients receiving clopidogrel plus aspirin compared with placebo plus aspirin, primarily gastrointestinal and at puncture sites. The incidence of ICH (0.1%) and fatal bleeding (0.2%) were the same in both groups. In CREDO, CLARITY, and COMMIT, there were no significant differences in major bleeding between patients receiving clopidogrel plus aspirin compared with placebo plus aspirin.71-73
Other adverse effects include diarrhea, nausea, and vomiting, which are common with ticlopidine and may occur in up to 30% to 50% of patients.97 Skin rash occurs rarely.98 Neutropenia is a serious side effect, and the incidence associated with ticlopidine is 1.3% to 2.1% as compared with 0.10% with clopidogrel.99,100 Neutropenia can be severe (<450 neutrophils/μL in 0.9% of patients treated with ticlopidine) and occasionally fatal.101 In the CAPRIE trial, the neutrophil count fell below 450 neutrophils/μL for 5 (0.05%) and 4 (0.04%) patients in the clopidogrel and aspirin groups, respectively.78 With ticlopidine, most cases develop within the first 3 months of therapy and initially may be clinically silent. Therefore, complete blood counts should be performed every 2 weeks during the first 3 months of therapy.102 Bone marrow aplasia and thrombotic thrombocytopenia purpura have been reported with ticlopidine, with an estimated incidence of 1 per 1,600 to 5,000 patients treated; this incidence is lower with clopidogrel.103,104 Ticlopidine had been reported to increase serum cholesterol by an average of 9%,105 but no change was associated with clopidogrel in the CAPRIE trial. Although hematologic complications may occur with clopidogrel (hemolytic uremic syndrome and thrombotic thrombocytopenic purpura), they appear to be rare.106 Overall, allergic or hematologic reactions occur in approximately 1% of patients taking clopidogrel, which is the thienopyridine of choice. Limited information on switching thienopyridine in patients with adverse reactions is available. In a recent study composed of 76 patients with an allergic or hematologic adverse reaction to clopidogrel or ticlopidine who had also received the other thienopyridine, 14 patients (27%) who had an allergic or hematologic adverse reactions to clopidogrel had a similar reaction (most commonly rash) to ticlopidine; none developed a life-threatening reaction.107 Desensitization protocols using escalating doses of oral clopidogrel have been proposed in allergic patients.108
The GP IIb/IIIa receptor is an integrin that mediates the final common pathway of platelet aggregation.109 There are approximately 80,000 receptors on the platelet surface.110 Integrins are heterodimers consisting of noncovalently associated α- and β-subunits.111 The GP IIb/IIIa receptor consists of the α IIb and β 3 subunits. The α-subunit is a 136-kDa molecule with a light and heavy chain; the light chain contains a short cytoplasmic tail, a transmembrane region, and a short extracellular domain, whereas the heavy chain is entirely extracellular.112 The β-subunit is a 84.5-kDa molecule with a short intracellular tail, transmembrane region, and a large extracellular domain.113 Platelet activation leads to a conformational change in the GP IIb/IIIa receptor, markedly increasing its affinity for its ligands through its binding sites.114 There are two main binding sites on the GP IIb/IIIa receptor. One recognizes the amino acid sequence arginine-glycine-aspartic acid (Arg-Gly-Asp or RGD) that is found on multiple ligands (fibronectin, von Willebrand factor [vWF], and vitronectin) but most importantly on fibrinogen, the major GP IIb/IIIa ligand, in which the RGD sequence occurs twice.115 The other peptide sequence is the Lys-Gln-Ala-Gly-Asp-Val, which is only located at the carboxyl terminus of the γ chain of fibrinogen.116 By competing with fibrinogen and vWF for GP IIb/IIIa binding, GP IIb/IIIa antagonists interfere with platelet aggregation.117 Because the GP IIb/IIIa receptor represents the final common pathway leading to platelet aggregation, these agents are more effective than other antiplatelet agents, such as aspirin and clopidogrel, in inhibiting platelets.
Currently, there are three parenteral GP IIb/IIIa antagonists in clinical use: abciximab, eptifibatide, and tirofiban. These agents are indicated only in patients with ACS undergoing PCI. Therefore, they are only administered within the hospital setting and are not used in the long-term care of patients with atherothrombotic vascular disease. Both preclinical and clinical pharmacodynamic studies have set the range of greater than 80% inhibition of platelet aggregation by light transmission aggregometry as the target for clinically effective antiplatelet activity.118 The degree of platelet inhibition appears central to the efficacy of GP IIb/IIIa inhibitors; achieving >95% platelet inhibition 10 minutes after the bolus in patients undergoing PCI was associated with a 55% reduction in major adverse cardiac events compared with those patients with <95% platelet inhibition.119 Although GP IIb/IIIa inhibitors have been shown to reduce major adverse cardiac events (death, MI, and urgent revascularization) by 35% to 50% in patients undergoing PCI, their broad use has been limited, as they are associated with an increased risk of bleeding complications.120
Abciximab is a large chimeric monoclonal antibody with a high binding affinity that translates into a prolonged pharmacologic effect.121,122 In particular, it is a monoclonal antibody that is a Fab (fragment-antigen binding) fragment of a chimeric human-mouse genetic reconstruction of 7E3. The Fc portion of the antibody was removed to decrease immunogenicity, and the Fab portion was attached to the constant regions of a human immunoglobulin. Abciximab has a high affinity for its receptor. Abciximab binding is specific for the β3-subunit and explains its ability to bind other β3-receptors. It has an almost equal potency for inhibition of the vitronectin (αVβ3) receptor and a lower affinity for cross-reacting with Mac-1 receptor (CD11b/CD18, αMβ2) found on leukocytes.123 Unlike the small-molecule GP IIb/III inhibitors, abciximab interacts with the GP IIb/IIIa receptor at sites distinct from the ligand-binding RGD sequence site, and exerts its inhibitory effect noncompetitively.124 Its plasma half-life is biphasic, with an initial half-life of less than 10 minutes and a second-phase half-life of approximately 30 minutes.125 However, because of its high affinity for the receptor, it has a biological half-life of 12 to 24 minutes, and because of its slow clearance from the body, it has a functional half-life up to 7 days.126 Because of the high affinity of abciximab for GP IIb/IIIa, the number of abciximab molecules bound to platelets is considerably higher than the free plasma pool of the drug for the duration of treatment, and platelet-associated abciximab can be detected for more than 14 days after the infusion is stopped.125 With an average period of platelet circulation of approximately 7 days, it appears that abciximab molecules can freely dissociate and reassociate with GP IIb/IIIa as the turnover of platelets in the circulation continues. The recommended dose for abciximab is 0.25 mg/kg bolus followed by an IV infusion of 0.125 μg/kg/min for 12 hours. No renal adjustments are required.
Eptifibatide and tirofiban are also known as small-molecule agents. Contrary to abciximab, these agents do not induce immune response and have lower affinity for the GP IIb/IIIa receptor. The stoichiometry of both eptifibatide and tirofiban is >100 molecules of drug per GP IIb/IIIa receptor needed to achieve full platelet inhibition. This compares with a stoichiometry of 1.5 molecules of abciximab for each receptor.125 Eptifibatide is a small, reversible, and highly selective heptapeptide with a rapid onset and a short plasma half-life (2-2.5 hours).127 Its molecular design is based on barbourin, a 73-amino acid peptide isolated from the venom of the Southeastern pygmy rattlesnake Sistrurus m barbourin.128 This is a unique member of the disintegrin family that contains a novel Lys-Gly-Asp (KGD) sequence, making it highly specific for the GP IIb/IIIa receptor. Plasma concentration of eptifibatide is proportional to the administered dose. Approximately 25% of eptifibatide of the drug molecules in plasma are protein bound, leaving the remaining 75% to constitute the pool of pharmacologically active drug.125 The recommended dose for eptifibatide depends on its indication for use. The initial bolus treatment ranges from 135 μg/kg when administered for the treatment of an ACS to a double bolus of 180 μg/kg when administered for PCI. Accordingly, the recommended continuous infusion dosing regimen ranges from 0.5 μg/kg/min to 2.0 μg/kg/min for an ACS (for up to 72 hours) versus PCI (for a minimum of 12 hours), respectively. With the recommended bolus (180 μg/kg followed by second 180 μg/kg bolus) and infusion (2 μg/kg/min) regimen, peak plasma levels are established shortly after the bolus dose, and slightly lower concentration are subsequently maintained throughout the infusion period. Plasma concentration decreases rapidly after the infusion is discontinued. Because the majority of the drug is eliminated via the kidney, a lower infusion dose (1 μg/kg/min) of eptifibatide is recommended in patients with creatinine clearance less than 50 mL/min. Substantial recovery of platelet aggregation is apparent within 4 hours of completion of infusion, whereas bleeding times returns to baseline within 1 hour.125
Tirofiban is a tyrosine-derived nonpeptide inhibitor that functions as a mimic of the RGD sequence and is highly specific for the GP IIb/IIIa receptor.129 Tirofiban is associated with a rapid onset and short duration of action, with a plasma half-life of approximately 2 hours.118 Like eptifibatide, substantial recovery of platelet aggregation is apparent within 4 hours of completion of infusion.125 The recommended dosing regimen for an ACS is a bolus 0.4 μg/kg/min for 30 minutes followed by infusion 0.10 μg/kg/min. Because the majority of the drug is eliminated through renal mechanisms, doses need adjustments for patients with renal insufficiency starting at a creatinine clearance of 30 mL/min (bolus 0.2 μg/kg/min for 30 minutes followed by infusion 0.05 μg/kg/min). It is not currently approved in the United States for PCI, although it is both approved and widely used throughout Europe for this indication (bolus 10 μg/kg followed by infusion 0.15 μg/kg/min for 18-24 hours). Several studies have documented that this approved bolus and infusion regimen for tirofiban achieves suboptimal levels of platelet inhibition for up to 4 to 6 hours that likely accounted for inferior clinical results in the PCI setting.130
Before the era of pretreatment with high loading doses of clopidogrel, the safety and efficacy of GP IIb/IIIa inhibition was tested in a large number of clinical studies that included patients with ACS as well as stable CAD. The landmark trial demonstrating efficacy of GP IIb/IIIa inhibition in the PCI setting was the Evaluation of IIb/IIIa Platelet Receptor Antagonist 7E3 in Preventing Ischemic Complications (EPIC) trial.131 In this study, high-risk patients undergoing balloon angioplasty were randomized to abciximab bolus and infusion versus abciximab bolus alone versus placebo. The group treated with abciximab bolus and infusion had a 35% lower rate of death, MI, or unplanned urgent revascularization at 30 days compared with the placebo group (8.3% vs 12.8%; P = .008). No significant benefit with abciximab bolus alone was observed, suggesting that shorter duration of platelet inhibition was insufficient to favorably affect clinical outcomes. A significant reduction in the primary end point with abciximab was also observed up to 3 years.132 Major bleeding complications occurred in a high proportion of patients treated with abciximab compared with placebo (major bleeding 14% vs 7%, transfusion 15% vs 7%, respectively). A series of procedural modifications, including performing front-wall arterial access only, reducing arterial sheath size (from 8-Fr to 6-Fr), reducing heparin dosing (target activated clotting time [ACT] 200-250 seconds rather than >300 seconds), removing sheaths as soon as possible (ACT <180 seconds) rather than overnight, and abandoning the use of routine venous sheaths, markedly reduced major bleeding complications to less than 1% to 1.5% in future trials.
After the EPIC trial, the Evaluation in PTCA to Improve Long-Term Outcome with Abciximab GP IIb/IIIa Blockade (EPILOG) trial was performed and also included patients undergoing balloon angioplasty but who were at a lower risk than patients in EPIC.133 In EPILOG, abciximab was given with two different lower doses of weight-adjusted heparin than had been administered in EPIC; a lower weight-adjusted infusion dose of abciximab was also implemented. This study was stopped prematurely due to efficacy with a significant reduction in the incidence of death and MI in patients treated with abciximab. Bleeding was lowest in patients who received the lower dose of heparin. Similar results were reported in the Evaluation of Platelet GP IIb/IIIa Inhibition in Stenting (EPISTENT) trial, which was the first randomized trial examining the use of GP IIb/IIIa inhibitors among patients undergoing stent placement.134 This trial randomized 2399 patients to stent plus placebo, stent plus abciximab, or balloon angioplasty plus abciximab. The primary 30-day end point, a combination of death, MI, or urgent revascularization, occurred in 10.8% of patients in the stent plus placebo group, 5.3% of those in the stent plus abciximab group (hazard ratio 0.48; P < .001), and 6.9% in the group undergoing balloon angioplasty plus abciximab (hazard ratio 0.63; P = .007). These benefits were maintained up to 1 year, with a significant reduction in 1-year mortality in patients treated with stent plus abciximab compared with stent without the IIb/IIIa inhibitor (2.4% vs 1.0%; P = .037).135 No significant differences in bleeding complications were noted among the various groups.
The first major trial to investigate eptifibatide was the Integrilin to Minimize Platelet Aggregation and Prevent Coronary Thrombosis-II (IMPACT-II) trial.136 In this trial, a significant reduction of ischemic events was found 24 hours after PCI. However, by 30 days, the difference was no longer statistically significant. Retrospective pharmacodynamic analysis of platelet function determined that a suboptimal dose of eptifibatide had been selected.137 The Enhanced Suppression of the Platelet IIb/IIIa Receptor with Integrilin (ESPRIT) trial therefore used a higher dose of eptifibatide than was used in IMPACT-II.138 The ESPRIT trial randomized 2064 patients undergoing stenting to eptifibatide (180 μg/kg bolus followed by a 2.0 mg/kg/h infusion with a second bolus of 180 μg/kg given 10 minutes after the first bolus) or placebo. In this trial, patients were administered a loading dose of clopidogrel or ticlopidine on the day of the procedure. The trial was terminated early for efficacy. The primary end point (composite of death, MI, urgent revascularization, or thrombotic bailout at 48 hours) was reduced by 37% with eptifibatide (10.5% vs 6.6%; P = .0017). These benefits were maintained at 6 months139 and up to 1 year.140 Major bleeding was rare, but occurred more frequently in patients receiving eptifibatide compared with placebo (1.3% vs 0.4%, respectively; P = .027).
On the basis of these trials, GP IIb/IIIa inhibitors became a cornerstone in treatment of patients undergoing PCI because of their ability to improve short- and long-term outcome, mostly by reducing the occurrence of periprocedural MI. Subsequently, however, it was shown that the benefits of GP IIb/IIIa blockade were reduced if patients were pretreated with a thienopyridine.141,142 In the first Intracoronary Stenting and Antithrombotic Regimen: Rapid Early Action for Coronary Treatment (ISAR-REACT) trial, low-to-intermediate risk patients (n = 2159) undergoing elective PCI, all of whom had been pretreated for at least 2 hours with a 600-mg loading dose of clopidogrel, were randomized to receive either abciximab therapy or placebo in a double-blind manner.143 The composite end point (death, MI, and urgent target vessel revascularization at 30 days) was similar in the two treatment groups (P = .82). The benefit was sustained at 1 year.144 Overall, these findings suggest that abciximab offers no further clinical benefit in low-to-intermediate risk patients scheduled for PCI if they have been pretreated with 600 mg of clopidogrel for at least 2 hours. The impact of adjunctive abciximab treatment in patients pretreated with a 600-mg loading dose of clopidogrel in an ACS setting was evaluated in the ISAR-REACT 2 trial.145
Diabetic patients with CAD who undergo PCI, particularly those requiring insulin, present a special group of patients characterized by a worse outcome after PCI due to an increased risk of both thrombosis and restenosis.146 A meta-analysis of six large trials evaluating the effect of GP IIb/IIIa inhibitors in ACS patients observed a 22% reduction of mortality at 30 days in diabetes mellitus patients (n = 6458) associated with the use of GP IIb/IIIa blockers compared with those not receiving these agents (4.6% vs 6.2%; P = .007), whereas nondiabetic patients (n = 23,072) had no benefit in survival.147 Of note, the benefit among diabetic patients was greater in those patients (n = 1279) who underwent PCI during the index hospitalization (1.2% vs 4%; P = .002). However, few patients included in these trials were adequately pretreated with a thienopyridine.
The Intracoronary Stenting and Antithrombotic Regimen: Is Abciximab a Superior Way to Eliminate Elevated Thrombotic Risk in Diabetics (ISAR-SWEET) trial was the first dedicated randomized trial evaluating GP IIb/IIIa blockade in diabetic patients scheduled for elective PCI.148 The trial was designed similarly to ISAR-REACT, and patients were randomized to treatment with abciximab or placebo after having all been pretreated with a 600-mg loading dose of clopidogrel. However, in this trial, abciximab did not reduce incidence of the composite end point (death or MI) at 30 days or 1 year (abciximab 8.3% vs placebo 8.6%; P = .91). Interestingly, angiographic restenosis and target vessel revascularization was significantly lower in the group of diabetic patients who received abciximab (P = .01 and P = .03, respectively). The impact of abciximab on the reduction of restenosis was subsequently investigated in the Intracoronary Stenting or Angioplasty for Restenosis in Small Arteries (ISAR-SMART) 2 trial, in which abciximab failed to reduce the incidence of angiographic restenosis after PCI of small coronary arteries.149 The inability of abciximab to reduce restenosis was also shown in other studies.150-152
The first trial to evaluate abciximab in patients with an ACS was the EPIC trial, in which only patients undergoing PCI were included and there was a 35% reduction in the combined end point for the group of patients treated with an abciximab bolus plus infusion.131 In the c7E3 Fab Antiplatelet Therapy in Unstable Refractory Angina (CAPTURE) trial, 1265 patients with refractory angina undergoing PCI were randomly assigned to receive abciximab 18 to 24 hours before PCI and for 1 hour after completion of the procedure or placebo.153 The primary end point, composite of death, MI, or urgent revascularization, was reduced in the abciximab group compared with placebo (15.9% vs 11.3%; P = .012). There was a significant increase in major bleeding (1.9% vs 3.8%; P = .043) and the need for transfusion (3.4% vs 7.1%; P = .005) in the abciximab group compared with placebo. Importantly, in a retrospective subgroup analysis of patients with an elevated and normal troponin T, it was demonstrated that only patients with an elevated troponin derived benefit from abciximab.154 The role of abciximab in ACS patients treated medically was investigated in the Global Use of Strategies to Open Occluded Arteries IV-Acute Coronary Syndrome Trial (GUSTO-IV ACS),155 in which 7,800 patients with UA/NSTEMI were randomized to receive placebo, abciximab for 24 hours, or abciximab for 48 hours. In this trial, PCI was prohibited by protocol for the first 60 hours after enrollment. This was the first study of abciximab that failed to show any benefit. In fact, not only was there no benefit, but there was a trend toward higher rates of MI and death with abciximab, which was greatest with the longer duration of treatment. Bleeding and thrombocytopenia were significantly increased by abciximab. Therefore, there is no evidence supporting the use of abciximab in ACS patients not undergoing PCI.
In the Platelet Glycoprotein IIb/IIIa in Unstable Angina: Receptor Suppression Using Integrilin Therapy (PURSUIT) trial, 10,948 patients with UA/NSTEMI were randomized to eptifibatide or placebo.156 A third arm with low-dose eptifibatide (1.3 μg/kg/min) was stopped prematurely by design after 3218 patients had been randomized, and the safety of the high-dose eptifibatide (2.0 μg/kg/min) arm was found to be acceptable. The primary end point of 30-day death or MI was reduced in those patients receiving eptifibatide versus placebo (14.2% vs 15.7%; P = .042). This treatment benefit was more pronounced among patients undergoing PCI within 72 hours of presentation (11.6% vs 16.7%; P = .01). Moderate or severe hemorrhage was more common in the eptifibatide group (12.8% vs 9.9%; P <.001).
Tirofiban was studied in the Platelet Receptor Inhibition in Ischemic Syndrome Management in Patients Limited by Unstable Signs and Symptoms (PRISM-PLUS) trial, in which 1915 patients with high-risk UA/NSTEMI were assigned to tirofiban, tirofiban plus heparin, or heparin alone.157 The combination of tirofiban and heparin led to a 32% risk reduction in the rate of death, MI, or recurrent refractory ischemia at 7 days compared with heparin alone (12.9% vs 17.9%; P = .004). The tirofiban treatment effect was also observed in patients (n = 475) undergoing PCI, with a 44% relative reduction in death or MI at 30 days.
Limited data are available on head-to-head comparisons of the different GP IIb/IIIa inhibitors. Two GP IIb/IIIa inhibitors, tirofiban and abciximab, were compared in the Do Tirofiban and ReoPro Give Similar Efficacy Outcomes (TARGET) trial, which randomized 5308 patients to tirofiban or abciximab before undergoing PCI with the intent to perform stenting.152 The primary end point was a composite of death, nonfatal MI, or urgent target vessel revascularization at 30 days. The primary end point (6.0% vs 7.6%; P = .038) as well as the incidence of MI (5.4% vs 6.9%; P = .04) were significantly lower in the abciximab group compared with the tirofiban group, respectively. There was no significant difference in the rate of major bleeding between the two groups; however, the incidence of minor bleeding and thrombocytopenia was significantly greater in the abciximab group. It was subsequently shown that the loading dose of tirofiban used in the trial was too small to inhibit platelet aggregation sufficiently in the crucial first 20 minutes after tirofiban had been given. In fact, the US Food and Drug Administration (FDA)–approved dosing regimens for abciximab and eptifibatide result in profound platelet inhibition (>80%) within 10 minutes of the bolus. In contrast, the dosing regimens for tirofiban in PRISM-PLUS (0.4 μg/kg/min load over 30 minutes followed by an infusion of 0.1 μg/kg/min for 12-24 hours thereafter) and in the TARGET (10 μg/kg bolus followed by an infusion of 0.15 μg/kg/min for 18-24 hours post PCI) led to inadequate platelet inhibition (60%-80%) up to 3 to 6 hours after the bolus, leading some investigators to recommend that the bolus dose for tirofiban may need to be increased 2.5- to 3-fold.158 Subsequent studies revealed that the loading dose had to be 2.5 times larger than was used in TARGET to provide the same degree of inhibition of aggregation as abciximab, with several small randomized clinical trials showing that tirofiban as effective as abciximab.159,160
Two different timing strategies for the administration of GP IIb/IIIa inhibitors have been used in the large randomized trials reviewed above: either early after the diagnosis of ACS before angiography (upstream treatment) or in the cardiac catheterization laboratory in patients about to undergo PCI. The optimal timing strategy of GP IIb/IIIa administration was assessed in the Acute Catheterization and Urgent Intervention Triage Strategy (ACUITY) Timing trial was performed, in which a total of 9207 ACS patients undergoing an invasive treatment strategy were randomly assigned to receive either routine upstream versus selective in-lab treatment with a GP IIb/IIIa inhibitor.161 All three GP IIb/IIIa inhibitors currently in clinical use (abciximab, tirofiban, and eptifibatide) could be used. The main result of the trial is that after 30 days, the routine upstream use of GP IIb/IIIa inhibitors in ACS patients with an invasive strategy produced a nonstatistically significant 12% decrease in the combined end point of death, MI, or target vessel revascularization. The difference (7.9% for in-lab vs 7.1% for upstream) did not meet the criterion for noninferiority. Importantly, the duration of upstream treatment was somewhat limited (~4 hours). Most recently, the Early Glycoprotein IIb/IIIa Inhibition in Non–ST-Segment Elevation Acute Coronary Syndrome (EARLY ACS) trial evaluated early administration of eptifibatide versus matching placebo infusion with provision eptifibatide after angiography (delayed eptifibatide) in 9492 patients with UA/NSTEMI undergoing invasive management.162 The primary end point occurred in 9.3% of patients in the early-eptifibatide group and in 10.0% in the delayed-eptifibatide group (P = .23). At 30 days, the rate of death or MI was 11.2% in the early-eptifibatide group, as compared with 12.3% in the delayed-eptifibatide group (P = .08). Patients in the early-eptifibatide group had significantly higher rates of bleeding and red-cell transfusion. A recent study suggests that eptifibatide infusion can be abbreviated safely to approximately 2 hours without adversely impacting ischemic outcomes (ie, noninferior) and is associated with less major bleeding.163 Overall, the findings of the above mentioned studies do not support the routine use of upstream compared with in-lab GP IIb/IIIa inhibition in ACS patients undergoing PCI.
Ultimately, these trials that favored GP IIb/IIIa inhibition in ACS patients undergoing PCI, particularly those patients who were troponin positive, were not designed to address the impact and value of GP IIb/IIIa inhibitors in the era of routine treatment with a high loading dose of clopidogrel before PCI. This was assessed in the ISAR-REACT 2 trial.145 The objective of ISAR-REACT 2 was to assess whether abciximab, administered in the catheterization laboratory, is associated with a clinical benefit in patients with an ACS undergoing PCI more than 2 hours after pretreatment with 600-mg loading dose of clopidogrel. In this double-blind randomized trial, 2,022 patients were included and assigned to receive either abciximab or placebo in addition to treatment with IV heparin, aspirin, and the 600-mg loading dose of clopidogrel. The study revealed that the administration of abciximab significantly reduced the incidence of the primary end point of death, MI, or target vessel revascularization at 30 days (relative risk, 0.75; 95% CI, 0.58-0.97; P = .03). The benefit of abciximab treatment, however, was restricted only to those patients who presented with an elevated troponin. Troponin-negative patients demonstrated substantially lower and almost identical event rates with abciximab versus placebo. Overall, based on the results of ISAR-REACT 2 trial and retrospective analysis of other randomized trials, these findings suggest that in the modern era of interventional cardiology using high clopidogrel dosing regimens, GP IIb/IIIa inhibition should be reserved only for high-risk ACS patients with positive cardiac markers.
The first large-scale trial to investigate the impact of abciximab treatment in STEMI patients treated with PCI was ReoPro and Primary PTCA Organization and Randomized Trial (RAPPORT), in which 483 patients with STEMI within 12 hours of presentation were randomized to placebo or abciximab during balloon angioplasty.164 The primary end point was a composite of death, reinfarction, or any target vessel revascularization at 6 months. The composite end point occurred 28.1% in the abciximab arm as compared with 28.2% in the placebo arm (P = .97). However, abciximab significantly reduced the incidence of death, reinfarction, or urgent target vessel revascularization at all time points assessed (9.9% vs 3.3%, P = .003, at 7 days; 11.2% vs 5.8%, P = .03, at 30 days; and 17.8% vs 11.6%, P = .05, at 6 months). Major bleeding occurred significantly more frequently in the abciximab group (16.6% vs 9.5%; P = .02), mostly at the arterial access site.
In the Abciximab Before Direct Angioplasty and Stenting in Myocardial Stenting Regarding Acute and Long-Term Follow-Up (ADMIRAL) trial, 300 patients with acute MI were randomized to abciximab plus stenting or stenting alone before they underwent coronary angiography.165 At 30 days, the primary end point (composite of death, reinfarction, or urgent target vessel revascularization) had occurred less in the abciximab compared with the placebo group (6.0% vs 14.6%; P = .01). This beneficial effect was sustained at 6 months (7.4% vs 15.9%; P = .02). Similar results were observed in the Intracoronary Stenting and Antithrombotic regimen-2 (ISAR-2) trial, in which 401 STEMI patients were randomized to receive either abciximab and reduced-dose heparin or full-dose heparin alone for PCI.166 Thirty days after PCI, the composite clinical end point of death, reinfarction, and target vessel revascularization was reached in 5.0% of the abciximab group versus 10.5% of the control group (P = .038). After 1 year of follow-up, the absolute reduction in the composite clinical end point with abciximab was still 5.7%, although the difference was no longer statistically significant.
The largest trial investigating the impact of GP IIb/IIIa inhibition in STEMI patients was the Controlled Abciximab and Device Investigation to Lower Late Angioplasty Complication (CADILLAC) trial, in which 2082 patients with acute MI were randomized in a 2 × 2 factorial design to balloon alone, balloon plus abciximab, stenting alone, or stenting plus abciximab.167 The primary end point was a composite of death from any cause, reinfarction, repeated intervention or revascularization of the target vessel as a result of ischemia, or disabling stroke during the first 6 months after the index procedure. The composite end point occurred in 8.3% in balloon alone group, 4.8% in balloon plus abciximab, 5.7% in stenting alone, and 4.4% in stenting plus abciximab (P = .02).
The value of abciximab in STEMI patients was evaluated in a large meta-analysis of randomized trials indicating substantial benefits from abciximab in STEMI patients treated with PCI.168 In this meta-analysis, a total of 11 trials in which 27,115 patients had been enrolled were analyzed. The meta-analysis demonstrated that the administration of abciximab to STEMI patients was associated with a significant reduction in 30-day (2.4% vs 3.4%; P = .047) and long-term (4.4% vs 6.2%; P = .01) mortality in patients treated with primary angioplasty. The frequency of reinfarction at 30 days was also significantly reduced by the administration of abciximab (2.1% vs 3.3%; P <.001).
All of the above studies evaluating GP IIb/IIIa inhibitors in STEMI were conducted in patients who had not been pretreated with clopidogrel. Whether there is added value from administering abciximab to patients suffering an STEMI and undergoing PCI after pretreatment with a high loading dose of clopidogrel was investigated in the third Bavarian Reperfusion Alternatives Evaluation (BRAVE-3) trial, in which a total of 800 patients with STEMI within 24 hours from symptom onset, all treated with 600 mg of clopidogrel, were randomly assigned in a double-blind fashion to receive either abciximab (n = 401) or placebo (n = 399) before being sent to the catheterization laboratory.169 The primary end point, infarct size measured by single-photon emission computed tomography with technetium-99m sestamibi before hospital discharge, was 15.7% ± 17.2% (mean ± SD) of the left ventricle in the abciximab group and 16.6% ± 18.6% of the left ventricle in the placebo group (P = .47). At 30 days, there were no differences in the composite of death, recurrent MI, stroke, or urgent revascularization of the infarct-related artery in the abciximab group (5.0%) and placebo group (3.8%) (P = .40). In conclusion, this study supports that upstream administration of abciximab is not associated with a reduction in infarct size in patients presenting with acute MI within 24 hours of symptom onset and receiving 600 mg of clopidogrel.
Costs have also been considered an issue when choosing GP IIb/IIIa inhibitor, with abciximab being more expensive than small-molecule inhibitors. Therefore, replacing abciximab with tirofiban, administered as a single high-dose bolus regimen, could represent a promising cost-saving strategy. The Single High Dose Bolus Tirofiban and Sirolimus Eluting Stent vs Abciximab and Bare Metal Stent in Myocardial Infarction (STRATEGY) trial evaluated the clinical and angiographic impact of single high-dose bolus tirofiban plus sirolimus-eluting stenting versus abciximab plus BMS in patients (n = 175) with STEMI.170 The cumulative incidence of death, reinfarction, stroke, or target vessel revascularization was significantly lower in the tirofiban plus sirolimus-eluting stent group (18%) versus the abciximab plus BMS group (32%) (hazard ratio, 0.53; P = .04), predominantly reflecting a reduction in the need for target vessel revascularization. The MULTI-STRATEGY trial was a 2 × 2 factorial design study that evaluated the effect of high-dose bolus tirofiban and of sirolimus-eluting stents as compared with abciximab infusion and uncoated-stent implantation in patients (n = 745) with STEMI undergoing PCI.171 ST-segment resolution occurred in 302 of 361 patients (83.6%) who had received abciximab infusion and 308 of 361 (85.3%) who had received tirofiban infusion (P <.001 for noninferiority). Ischemic and hemorrhagic outcomes were similar in the tirofiban and abciximab groups. At 8 months, major adverse cardiac events occurred in 14.5% with uncoated stents and 7.8% with sirolimus stents (P = .004), predominantly reflecting a reduction of revascularization rates. In conclusion, this study showed that in patients with STEMI undergoing primary PCI, compared with abciximab, tirofiban therapy was associated with noninferior resolution of ST-segment elevation at 90 minutes after coronary intervention, whereas sirolimus-eluting stent implantation was associated with a significantly lower risk of major adverse cardiac events than uncoated stents within 8 months after intervention.
Effective and rapid reperfusion of the infarct-related coronary artery, irrespective of the manner with which it is achieved, is the critical goal in the treatment of acute MI. The optimal pharmacologic strategy for bridging between admission and performance of PCI in a patient with acute MI has not been fully defined. Primary PCI is more effective than thrombolytic therapy for the treatment of STEMI.172 However, time to reperfusion is a critical determinant of outcome with both these strategies.173 Facilitated PCI offers both theoretical and practical appeal to overcome time limitations. To date, trials that have compared primary PCI with facilitated PCI with full-dose fibrinolytics have failed to show a clinical benefit, although infarct artery patency rates before the PCI were significantly higher with facilitated PCI.174 This has prompted the investigation of the potential benefits of other facilitated PCI strategies, which include the use of upstream GP IIb/IIIa inhibitors alone or in combination with a reduced-dose fibrinolytic.175
In the ADMIRAL study, patients who received their randomly assigned treatment with abciximab early had a greater benefit with respect to the primary end point at both 30 days and 6 months than did those treated with abciximab in the intensive care unit or catheterization laboratory, thereby demonstrating the potential benefit of a facilitated strategy.174 The investigators attributed the better outcome of abciximab to higher levels of TIMI grade 3 flow in the target vessel immediately before (16.8% vs 5.4%; P = .01) and immediately after (95.1% vs 86.7%; P = .04) the procedure.
The Integrilin in Acute Myocardial Infarction (INTAMI) trial evaluated adjunctive therapy with eptifibatide administered early in the emergency department (n = 53) compared with late, optional administration in the catheterization laboratory (n = 49).176 TIMI grade 3 flow at the time of angiography was higher in the early eptifibatide group compared with the late/no eptifibatide group (34.0% vs 10.2%; P = .01). The presence of visible thrombus also trended lower in the early group (57.7% vs 70.8%; P = .1). However, there was no difference in post-PCI TIMI flow grade 3, TIMI myocardial perfusion grade 3, ST-segment resolution, and clinical events or bleeding by 30 days. The Time to Integrilin Therapy in Acute Myocardial Infarction (TITAN–TIMI 34) trial compared a strategy of early initiation of eptifibatide in the emergency department (n = 174) with that of initiating eptifibatide in the cardiac catheterization laboratory (n = 142).177 The primary end point of corrected TIMI frame count on diagnostic angiography was lower in the emergency department group (77.5 frames vs 84.3 frames; P = .049). TIMI myocardial perfusion grade 3 was present more frequently in the emergency department group (24.3% vs 14.2%; P = .026).
Upstream administration of GP IIb/IIIa inhibitors before PCI in patients presenting with acute MI has been studied in three trials. In the Tirofiban Given in the Emergency Room Before Primary Angioplasty (TIGER-PA) Pilot Trial, 100 patients with acute MI were randomized to either administration of tirofiban in the emergency room or later administration in the catheterization laboratory.178 Angiographic analysis showed a statistically significant difference in initial TIMI grade 3 flow (32% vs 10%; P = .007), initial corrected TIMI frame counts (44 ± 20 vs 66 ± 23; P = .005), and TIMI grade 3 myocardial perfusion grade (32% vs 6%; P = .001). There were no differences noted between the two groups with regard to bleeding complications. This pilot study suggests that early administration of tirofiban improves angiographic outcomes without increasing the risk of bleeding. Similarly, eptifibatide was shown to improve TIMI grade 2 or 3 flow when given upstream to patients presenting with STEMI undergoing primary PCI compared with an historical control group of 30 patients receiving eptifibatide at the time of the PCI.179 The ongoing tirofiban in MI evaluation (ON-TIME) study randomized 507 patients within 6 hours of symptom onset to tirofiban started before transportation to a PCI center or tirofiban started on arrival at the PCI center. The trial showed a significant improvement in the number of patients with TIMI grade 2 or 3 flow for those who received upstream tirofiban compared with receiving tirofiban at the PCI center (43% vs 34%; P = .04).180 In the ON-TIME 2 study, 984 patients with STEMI who were candidates to undergo PCI were randomly assigned to either high-bolus dose tirofiban (n = 491) or placebo (n = 493) in addition to aspirin, heparin, and a 600-mg loading dose of clopidogrel. The primary end point was the extent of residual ST-segment deviation 1 hour after PCI, which was significantly lower in patients pretreated with high-bolus dose tirofiban (P = .003). The rate of major bleeding did not differ significantly between the two groups (P = .36).181
These small studies set the basis for larger studies to help clarify the safety and efficacy of different regimens of facilitated PCI using GP IIb/IIIa inhibitors alone or in combination with a reduced-dose fibrinolytic. In the Immediate Angioplasty Versus Standard therapy with Rescue Angioplasty After Thrombolysis in the Combined Abciximab Reteplase Stent Study in Acute Myocardial Infarction (CARESS-in-AMI) trial, 600 patients were treated with half-dose reteplase, abciximab, heparin, and aspirin and randomly assigned to immediate transfer to the nearest interventional facility for PCI or to management in the local hospital, with transfer only in case of persistent ST-segment elevation or clinical deterioration.182 Of the 299 patients assigned to immediate PCI, 289 (97.0%) underwent angiography, and 255 (85.6%) received PCI. Rescue PCI was done in 91 patients (30.3%) in the standard care/rescue PCI group. The primary outcome (composite of death, reinfarction, or refractory ischemia at 30 days) occurred in 4.4% in the immediate PCI group compared with 10.7% in the standard care/rescue PCI group (hazard ratio 0.40; log rank P = .004). There were no significant differences in major bleeding (3.4% vs 2.3%, respectively; P = .47). Overall, this trial demonstrates that immediate transfer for PCI improves outcome in high-risk patients with STEMI treated at a noninterventional center with half-dose reteplase and abciximab.
In the Facilitated Intervention With Enhanced Reperfusion Speed to Stop Events (FINESSE) trial, patients (n = 2452) with STEMI who presented ≤6 hours after the onset of symptoms were randomized to receive combination-facilitated PCI, abciximab-facilitated PCI, or primary PCI.183 All patients received heparin (unfractionated heparin or enoxaparin) before PCI and a 12-hour infusion of abciximab after PCI. The primary end point (composite of death from all causes, ventricular fibrillation occurring more than 48 hours after randomization, cardiogenic shock, and congestive heart failure during the first 90 days after randomization) occurred in 9.8%, 10.5%, and 10.7% of the patients in the combination-facilitated PCI group, abciximab-facilitated PCI group, and primary-PCI group, respectively (P = .55); 90-day mortality rates were 5.2%, 5.5%, and 4.5%, respectively (P = .49). Overall, the FINESSE study shows that neither facilitation of PCI with reteplase plus abciximab nor facilitation with abciximab alone significantly improved the clinical outcomes as compared with abciximab given at the time of PCI in patients with STEMI.
ACS trials tended to report higher incidence of thrombocytopenia compared with PCI trials, perhaps because of longer heparin infusions producing heparin-induced thrombocytopenia (HIT).184 In a meta-analysis,184 abciximab increased the incidence of mild thrombocytopenia (>50,000 <90,000-100,000) compared with placebo (4.2% vs 2.0%; P <.001; odds ratio [OR] 2.13). Patients receiving abciximab also had more than twice the incidence of severe thrombocytopenia (>20,000 and <50,000) than those receiving placebo (1.0% vs 0.4%; P = .01; OR 2.48). Eptifibatide or tirofiban did not increase mild or severe thrombocytopenia compared with placebo. Severe and profound (<20,000) thrombocytopenia are more commonly associated with abciximab use and require immediate cessation of therapy. An algorithm for evaluation of these patients has been proposed.185 Pseudothrombocytopenia secondary to platelet clumping and HIT needs to be ruled out. The platelet count usually returns to normal within 48 to 72 hours in most cases. Regardless of its etiology, thrombocytopenia in patients undergoing PCI is associated with more ischemic events, bleeding complications, and transfusions.186 The mechanism(s) of thrombocytopenia is unknown. The platelet count falls within hours of GP IIb/IIIa administration. Readministration of abciximab, but not the small-molecule inhibitors (eptifibatide and tirofiban), is associated with a slight increased risk of thrombocytopenia.187
| Class I |
1. Aspirin should be administered to UA/NSTEMI patients as soon as possible after hospital presentation and continued indefinitely in patients not known to be intolerant of that medication. (Level of Evidence: A) 2. Clopidogrel (loading dose followed by daily maintenance dose) should be administered to UA/NSTEMI patients who are unable to take ASA because of hypersensitivity or major gastrointestinal intolerance. (Level of Evidence: A) 3. In UA/NSTEMI patients with a history of gastrointestinal bleeding, when ASA and clopidogrel are administered alone or in combination, drugs to minimize the risk of recurrent gastrointestinal bleeding (eg, proton-pump inhibitors) should be prescribed concomitantly. (Level of Evidence: B) 4. For UA/NSTEMI patients in whom an initial invasive strategy is selected, antiplatelet therapy in addition to aspirin should be initiated before diagnostic angiography (upstream) with either clopidogrel (loading dose followed by daily maintenance dose) or an IV GP IIb/IIIa inhibitor. (Level of Evidence: A) Abciximab as the choice for upstream GP IIb/IIIa therapy is indicated only if there is no appreciable delay to angiography and PCI is likely to be performed; otherwise, IV eptifibatide or tirofiban is the preferred choice of GP IIb/IIIa inhibitor. (Level of Evidence: B) 5. For UA/NSTEMI patients in whom an initial conservative (ie, noninvasive) strategy is selected (see Section IV.C), clopidogrel (loading dose followed by daily maintenance dose) should be added to ASA and anticoagulant therapy as soon as possible after admission and administered for at least 1 month (Level of Evidence: A) and ideally up to 1 year. (Level of Evidence: B) |
| Class IIa |
1. For UA/NSTEMI patients in whom an initial conservative strategy is selected and who have recurrent ischemic discomfort with clopidogrel, ASA, and anticoagulant therapy, it is reasonable to add a GP IIb/IIIa antagonist before diagnostic angiography. (Level of Evidence: C) 2. For UA/NSTEMI patients in whom an initial invasive strategy is selected, it is reasonable to initiate antiplatelet therapy with both clopidogrel (loading dose followed by daily maintenance dose) and an IV GP IIb/IIIa inhibitor. (Level of Evidence: B) Abciximab as the choice for upstream GP IIb/IIIa therapy is indicated only if there is no appreciable delay to angiography and PCI is likely to be performed; otherwise, IV eptifibatide or tirofiban is the preferred choice of GP IIb/IIIa inhibitor. (Level of Evidence: B) 3. For UA/NSTEMI patients in whom an initial invasive strategy is selected, it is reasonable to omit upstream administration of an IV GP IIb/IIIa antagonist before diagnostic angiography if bivalirudin is selected as the anticoagulant and at least 300 mg of clopidogrel was administered at least 6 h earlier than planned catheterization or PCI. (Level of Evidence: B) |
| Class IIb |
| For UA/NSTEMI patients in whom an initial conservative (ie, noninvasive) strategy is selected, it may be reasonable to add eptifibatide or tirofiban to anticoagulant and oral antiplatelet therapy. (Level of Evidence: B) |
| Class III |
| Abciximab should not be administered to patients in whom PCI is not planned. (Level of Evidence: A) |
| GP IIb/IIIa Inhibitors |
| Class IIa |
| It is reasonable to start treatment with glycoprotein IIb/IIIa receptor antagonists (abciximab [Level of Evidence: A], tirofiban [Level of Evidence: B], or eptifibatide [Level of Evidence: B] at the time of primary PCI (with or without stenting) in selected patients with STEMI. |
| Class IIb |
| The usefulness of glycoprotein IIb/IIIa receptor antagonists (as part of a preparatory pharmacologic strategy for patients with STEMI before their arrival in the cardiac catheterization laboratory for angiography and PCI) is uncertain. (Level of Evidence: B) |
| Thienopyridines |
| Class I |
| 1. A loading dose of thienopyridine is recommended for STEMI patients for whom PCI is planned. Regimens should be one of the following: |
| a. At least 300-600 mg of clopidogrel should be given as early as possible before or at the time of primary or nonprimary PCI. (Level of Evidence: C) |
| b. Prasugrel 60 mg should be given as soon as possible for primary PCI. (Level of Evidence: B) |
| c. For STEMI patients undergoing nonprimary PCI, the following regimens are recommended: |
| (i) If the patient has received fibrinolytic therapy and has been given clopidogrel, clopidogrel should be continued as the thienopyridine of choice. (Level of Evidence: C) |
| (ii) If the patient has received fibrinolytic therapy without a thienopyridine, a loading dose of 300 to 600 mg of clopidogrel should be given as the thienopyridine of choice. (Level of Evidence: C) |
| (iii) If the patient did not receive fibrinolytic therapy, either a loading dose of 300 to 600 mg of clopidogrel should be given, or once the coronary anatomy is known and PCI is planned, a loading dose of 60 mg of prasugrel should be given promptly and no later than 1 h after the PCI. (Level of Evidence: B) |
| 2. The duration of thienopyridine therapy should be as follows: |
| a. In patients receiving a stent (BMS or DES) during PCI for ACS, clopidogrel 75 mg daily (Level of Evidence: B) or prasugrel 10 mg daily (Level of Evidence: B) should be given for at least 12 months. |
| b. If the risk of morbidity because of bleeding outweighs the anticipated benefit afforded by thienopyridine therapy, earlier discontinuation should be considered. (Level of Evidence: C) |
| 3. In patients taking a thienopyridine in whom CABG is planned and can be delayed, it is recommended that the drug be discontinued to allow for dissipation of the antiplatelet effect. (Level of Evidence: C) The period of withdrawal should be at least 5 days in patients receiving clopidogrel (Level of Evidence: B) and at least 7 days in patients receiving prasugrel (Level of Evidence: C), unless the need for revascularization and/or the net benefit of the thienopyridine outweighs the potential risks of excess bleeding. (Level of Evidence: C) |
| Class IIb |
| 1. Continuation of clopidogrel or prasugrel beyond 15 months may be considered in patients undergoing DES placement. (Level of Evidence: C) |
Investigations of oral GP IIb/IIIa inhibitors, including sibrafiban, orbofiban, xemilofiban, and lotrafiban, have been halted due to negative results from several large trials in patients with ACS or undergoing PCI.188-191 Meta-analysis of these trials further demonstrated an increased bleeding risk and increased mortality in patients treated with oral GP IIb/IIIa inhibitors.192,193 One of the possible explanations for the poor outcomes and increased mortality in these trials was the interindividual variability in the level of platelet inhibition over time.194 Another potential mechanism is that partial-agonist properties of oral GP IIb/IIIa inhibitors enhance platelet activation, especially at times when the serum drug levels are low, leaving more GP IIb/IIIa receptors available for binding fibrinogen. Proinflammatory effects have also been considered as an explanation for the adverse events with oral GP IIb/IIIa inhibitors.195 Currently, only parenteral GP IIb/IIIa inhibitors are approved for clinical use.
Despite the use of what is generally regarded as “optimal” antiplatelet therapy, recurrent atherothrombotic events occur in some patients because of variability of individual response to treatment, which may lead in some patients to what is commonly termed as “resistance.”196 More strictly, resistance is a laboratory finding that consists in failure of an antiplatelet agent to block its specific target. Thus for aspirin resistance involves inadequate or lack of inhibition of the COX-1–mediated TXA2 pathway, whereas for clopidogrel, resistance involves P2Y12 receptor signaling. However, thrombotic events involve multiple signaling pathways. Therefore, it is inaccurate to ascribe an adverse outcome to drug resistance without actually performing laboratory testing. Without such confirmation, it should be considered treatment failure rather than resistance to a particular antiplatelet agent.197
Clinical studies have shown that 10% to 20% of patients with an atherothrombotic episode who were treated with aspirin had another event, which suggests that not all patients benefit equally from aspirin therapy and that variable interindividual response profiles may exist. The possibility of variable response to aspirin had been raised as far back as 1965 in a study of its effects on bleeding times.198 Untreated subjects (n = 10) as well as subjects treated with 650 or 1,350 mg of aspirin exhibited large interindividual differences in bleeding times. In another early report, it was shown that patients with elevated degrees of platelet reactivity while receiving aspirin therapy are at greater risk of ischemic events 5 years after recovery from an acute coronary event.199
A number of subsequent clinical studies have correlated aspirin resistance with long-term clinical outcomes, not only in patients with CAD, but also in patients with ischemic stroke or peripheral arterial disease.200-205 The prevalence of resistance reported varies considerably. The contrasting estimates reported in laboratory studies are largely assay-dependent. In many of these studies, assays that are not specific to COX-1 signaling were used. In fact, although many available platelet function tests are able to detect aspirin-induced effects, the results obtained are not all specific to the degree of COX-1 inhibition and may be reflective of other platelet-signaling pathways. However, when tests specifically assess COX-1 activity, aspirin produces resistance only infrequently (<5%), and the most common reason for exaggerated reports of aspirin resistance when using assays that specifically assess COX-1 activity is poor patient compliance.206,207 Further, the overall prevalence of inadequate aspirin effects may be influenced by the patient population under investigation. Patients with diabetes and ACS, typically characterized by hyperreactivity, also present with high platelet reactivity while receiving aspirin therapy. Genetic factors as well have been implied in reduced aspirin sensitivity.208 Interactions with drugs, such as ibuprofen, which interfere with aspirin-induced COX-1 acetylation, may also be a cause of inadequate or no effects of aspirin. The latter may also be responsible for an increased risk of ischemic effects despite aspirin use.209
Given that the primary cause of aspirin resistance is poor compliance to medication,208,209 it is imperative when interpreting a platelet function test assessing for aspirin responsiveness that the patient is compliant with treatment. The second most important factor in the management of aspirin-resistant patients is whether the patients are receiving treatment with substances, such as ibuprofen, which interfere with COX-1 acetylation. Some investigators have considered increasing the dose of aspirin in “resistant” patients. However, increasing the dose of aspirin has not been associated with an increase in COX-1 inhibition, as assessed by COX-1 specific assays. Of note, in vitro studies have shown that the dose of aspirin necessary to inhibit COX-1 is markedly lower than that used in clinical practice.18,19 Increasing the dose of aspirin has been associated with greater platelet inhibition, as assessed by assays nonspecific to COX-1.210 However, there is no evidence that increasing the dose of aspirin based on these measures is accompanied by better clinical outcomes. On the contrary, higher aspirin doses are known to be associated with an increased risk of bleeding.18,19 Although the meaning of different assays testing aspirin sensitivity (specific and nonspecific for COX-1 inhibition) is currently being better evaluated, several studies, including meta-analyses, using various laboratory assays support the poor prognostic implications of inadequate aspirin-induced effects (Fig. 61–3).211-221
Figure 61–3.
Risk of any cardiovascular event in aspirin-resistant patients. Twenty studies totaling 2930 patients with cardiovascular disease and treated with aspirin 75 to 325 mg were included. Aspirin-resistant compared with aspirin-sensitive patients were at a greater risk of any cardiovascular event (odds ratio 3.85, 95% CI, 3.08-4.80). Reproduced with permission from Krasopoulos et al.212
Accumulating data show that the problem of variability in individual responsiveness to antiplatelet therapy also applies to clopidogrel.222,223 However, the nature of variability of clopidogrel responsiveness is different compared with aspirin due to their pharmacologic profiles. The parent compound, clopidogrel bisulfate, is an inert prodrug that is biotransformed in the liver into its active metabolite by the CYP system. However, 85% of the prodrug is hydrolyzed into an inactive form, and only 15% is available to become an active agent through a double oxidation step in the liver. Blockade of the P2Y12 receptor by the active metabolite elicits a cascade of events that culminate in platelet inhibition. Indeed, each of these steps is subject to variability, which ultimately lead to variability in interindividual response profiles.
The occurrence of ischemic events despite the use of clopidogrel suggests that inadequate response to treatment may be present in some patients. This risk may be further enhanced in patients who have inadequate response to both aspirin and clopidogrel therapy.223 Similarly to aspirin, criteria to define inadequate response or “resistance” to clopidogrel have not been standardized. Obstacles to standardization include the multiplicity of assays available for measuring platelet function, differing ways in which laboratories use these assays, and lack of a standard definition for nonresponsiveness (Table 61–2).209 Turbidometric light transmittance aggregometry using ADP as the challenging agonist is still considered the gold standard to assess antiplatelet effects. Nonresponder has been defined as an absolute difference of ≤10% between baseline aggregation and posttreatment aggregation. Alternatively, clopidogrel responsiveness can be defined as the percent decrease in platelet aggregation or inhibition of platelet aggregation after treatment compared with baseline values.224,225 Inhibition of platelet aggregation values of <10%, 10% to 30%, and >30% have been used to define patients as nonresponders, low responders, and responders, respectively. Each definition may result in different values for responsiveness. However, it is now appreciated that observations of clopidogrel response that rely on baseline platelet reactivity are less reliable predictors of ischemic risk than posttreatment platelet reactivity.224,225 Table 61–3 summarizes the studies in which various measures of clopidogrel responsiveness, mainly posttreatment platelet reactivity, have been associated with adverse ischemic events, including stent thrombosis.66,221,224-245
| Aspirin |
|
|
|
| Clopidogrel |
|
|
| Population Studied | Clopidogrel Dose | Definition of Nonresponsiveness | Incidence of Nonresponsiveness | Adverse Clinical Events |
|---|---|---|---|---|
STEMI patients (n = 60) Ref. 224 | LD 300 mg MD 75 mg | LTA (5 μmol/L ADP); 1st quartile of reduction in aggregate size | 25% | 40% vs 6.7% (P for trend = .007) had ACS, acute peripheral arterial occlusion, mortality from ischemic stroke |
Patients undergoing nonemergent coronary stenting (n = 192) Ref. 225 | LD 300 mg (n = 75), 600 mg (n = 60) MD 75 mg | LTA (20 μmol/L ADP) | Not defined | Posttreatment aggregation 63% ± 12% in patients with events (CV death, MI, UA, or stroke at 6 mo) vs 56% ± 15% in patients without events (P = .02) |
Patients undergoing elective coronary stenting (n = 120) Ref. 226 | LD 300 mg (n = 60), 600 mg (n = 60) MD 75 mg | LTA (5 and 20 μmol/L ADP) | Not defined | Myocardial infarction only occurred in those patients with mean 5 μmol/L ADP-induced aggregation ≥50% |
Patients undergoing coronary stenting for UA/NSTEMI (n = 106) Ref. 228 | LD 300 mg MD 75 mg (n = 85), | LTA (10 μmol/L ADP); 4th quartile of aggregation | 22% | 22.4-fold increased risk of CV death, stent thrombosis, ischemic stroke, or recurrent ACS at 1 mo |
Patients undergoing elective coronary stenting (n = 120) Ref. 221 | 75 mg >5 d (n = 21) LD 300 mg MD 75 mg | LTA (5 and 20 μmol/L ADP); <10% reduction compared with baseline | 24% | 2.3-fold increased risk of creatine kinase-MB elevation after PCI (P = .06) |
Patients undergoing coronary stenting for UA/NSTEMI (n = 292) Ref. 227 | LD 300 mg (n = 146), 600 mg (n = 146) MD 75 mg | LTA (10 μmol/L ADP); aggregation >70% | LD 600 mg 15% LD 300 mg 25% | 13.8-fold increased risk of CV death, stent thrombosis, ischemic stroke, or recurrent ACS (P = .0001) at 1 mo |
Patients undergoing elective coronary stenting (n = 802) (EXCELSIOR) Ref. 228 | LD 600 mg MD 75 | LTA (5 μmol/L ADP); aggregation >14% (median value) | 50% | 6.7-fold risk of death, MI, and target lesion revascularization at 30 d for platelet aggregation >median (P = .003) |
Symptomatic CAD patients undergoing coronary stenting (n = 379) Ref. 229 | LD 600 mg MD 75 mg | LTA (20 μmol/L ADP); aggregation >70% | 5.8% | 3.7-fold increased risk of CV death, MI, or stroke at 3 mo |
Patients undergoing nonurgent coronary stenting (n = 100) Ref. 230 | 75 mg daily ≥1 mo | LTA (5 μmol/L ADP) aggregation >50% | 22% | 35-fold increased risk of CV death, MI, UA, TVR, non-TVR at 12 mo |
Patients undergoing coronary stenting for UA/NSTEMI (n = 190) Ref. 231 | LD 600 mg MD 75 mg | LTA (10 μmol/L ADP); aggregation >70% | 22% | 2.4-fold increased risk of troponin I elevation after PCI |
Type 2 diabetic patients (n = 173) 6-9 mo after coronary stenting Ref. 232 | 75 mg daily for 3-6 mo | LTA (20 μmol/L ADP); 4th quartile of aggregation | 26% | 3.4-fold increased risk of CV death, ACS, or stroke at 2 y |
Patients undergoing coronary stenting for STEMI (n = 367) Ref. 233 | LD 300 mg MD 75 mg | LTA (2 and 10 μmol/L ADP); ≥ 90th percentile of controls (20% and 70%, respectively) | 4.6% (2 μmol/L ADP) | CK-MB (P < .0001) and troponin I (P < .005) peak values in patients with nonresponse were significantly higher than in responders defined by LTA and PFA-100 |
| A point-of-care assay (PFA-100) (CT/EPI); <90th percentile of controls (203 s) | 23.9% (10 μmol/L ADP) | |||
| 32.9% (PFA-100) | ||||
Patients undergoing elective coronary stenting (n = 105) Ref. 234 | LD 600 mg MD 75 mg | LTA (5 and 20 μmol/L ADP); <10% reduction compared with baseline | 5%-11% | Subacute stent thrombosis occurred in two patients who were nonresponders after PCI. |
Patients undergoing urgent and nonurgent DES (n = 804) Ref. 235 | LD 600 mg MD 75 mg | LTA (10 μmol/L ADP) >70% Aggregation | 13% | 3.1-fold increased risk Definite/probable stent thrombosis |
Patients undergoing coronary stenting for stable angina and low-risk UA/NSTEMI (n = 144) Ref. 236 | LD 300 mg MD 75 mg | VASP-P assay; PRI > 50% | 80% | 0% vs 21% (P < .01) had MACE during the 6-mo (1st quintile vs upper quintiles) With this cut-off value, the assay had 100% sensitivity and 25% specificity for prediction of MACE. The area under the curve was 0.55 |
Patients undergoing coronary stenting for UA/NSTEMI (n = 195) Ref. 237 | LD 600 mg MD 75 mg | LTA (10 μmol/L ADP); ≥70% VASP-P assay; PRI >53% | 28% (LTA) 54% (VASP-P) | LTA; 20% vs 2.5% had CV event during the first month after PCI VASP-P; 12.3% vs 1.1% had CV event during the first month after PCI |
Patients with (n = 16) and without coronary SAT (n = 30) Ref. 238 | Ticlopidine 2×250 mg/d or clopidogrel 2×75 mg/d | VASP-P assay; PRI >50% | SAT group; 94% Without SAT group; not defined | 94% of patients with SAT had nonresponse |
Patients with (n = 30) and without coronary SAT (n = 100) Ref. 239 | MD 75 mg | LTA (5 and 20 μmol/L ADP); 4th quartile of aggregation VASP-P; 4th quartile of PRI | 60%-65% (LTA) 50% (VASP-P) in SAT patients | The SAT patients had higher mean platelet reactivity than those without SAT by all measurements (P < .05): 49% vs 33% for 5 μmol/L ADP and 65% vs 51% for 20 μmol/L ADP (P < .001), 69% vs 46% for PRI (P < .03) |
Patients undergoing urgent or elective coronary stenting for higher incidence of stent thrombosis (n = 99) Ref. 240 | LD 600 mg MD 75 mg | VASP-P assay; PRI >48% | Not defined | A cut-off value >48% for PRI had a high predictive value for SAT with a sensitivity of 80% and a specificity of 73% (area under the ROC curve 0.792; 95% confidence interval [CI] 0.689-0.874) |
Patients undergoing elective coronary stenting (n = 380) Ref. 241 | LD 600 mg or 75 mg daily ≥5 d | Point-of-care platelet aggregation assay; PRU ≥235 | 32% | 6.5% vs 1.0% (P = .008) had CV death, MI, or stent thrombosis at 6 mo |
Patients undergoing nonurgent coronary stenting (n = 160) (ARMYDA-PRO) Ref. 242 | LD 600 mg or 75 mg daily ≥5 d | Point-of-care platelet aggregation assay; PRU in the 4th quartile | 25% | 6-fold increased risk of cardiac death, MI, or TVR |
Patients undergoing coronary stenting for ACS (n = 683) Ref. 243 | LD 600 mg MD 75 mg | Point-of-care platelet aggregation assay; PRU ≥240 | 32% | 2.55-fold increased risk of CV death, 3.36-fold increased risk of nonfatal MI |
Patients undergoing urgent and nonurgent coronary stenting (n = 1,608) Ref. 244 | LD 600 mg MD 150 mg for 3 d then 75 mg daily | Point-of-care platelet aggregation assay; upper quintile | 20% | 11-fold increased risk of definite stent thrombosis at 30 d |
Patients with (n = 10) and without SAT (n = 22), healthy volunteers (n = 17) Ref. 245 | LD 300 mg | LTA (3.2 μmol/L ADP); >45% | 22% (LTA) | Nonresponder defined as LTA was 30% in SAT group, 18% in without SAT group (P = NS). SIPA was significantly higher in SAT group than in without SAT group both at 200 s−1 (P = .0008) and 4000 s−1 (P = .0002). |
| MD 75 mg | SIPA; not defined | Not defined (SIPA) |
The mechanisms underlying interindividual variability in response to clopidogrel have not been defined but are probably multifactorial and include genetic, cellular, and clinical factors.224,225 Among the genetic factors, various polymorphisms have been studied. Among these, genetic polymorphisms of CYP enzymes, which are implied in generating the active metabolite of clopidogrel, appear to play a more important role than downstream targets, such as platelet membrane receptors. Among the genetic variations of CYP enzymes, variant alleles of 2C19 appear to be the most important, as demonstrated by pharmacokinetic, pharmacodynamic, and clinical outcome studies.246,247 Cellular factors may also play a role in clopidogrel response variability. These include more rapid platelet turnover, increased platelet exposure to ADP, reduced CYP activity, upregulation of purinergic signaling (P2Y1 and P2Y12), and the upregulation of nonpurinergic pathways.224,225 Clinical factors have shown to have an important role in clopidogrel response. Similar to aspirin, failure to prescribe/poor compliance play a pivotal role. Certain clinical scenarios, such as diabetes, ACS, and elevated body mass index, are also associated with reduced clopidogrel effects.248-253 This may contribute to why these patients have a greater likelihood of developing recurrent thrombotic complications despite clopidogrel use. Drug-drug interactions, including lipophilic statins254 and omeprazole,255 which are metabolized by the CYP system, may interfere with the metabolism of clopidogrel and thus impact its pharmacokinetic/pharmacodynamic effects. However, to date there is conflicting evidence that these drug-drug interactions have clinical impact.256,257
Due to its potentially severe consequences, inadequate clopidogrel effects has emerged an important clinical problem warranting measures to overcome this phenomenon. The first and foremost important approach is to ensure patient compliance. Although ongoing studies will better define drug-drug interactions (eg, clopidogrel-omeprazole interaction), this needs to be ruled out as a potential cause of inadequate clopidogrel responsiveness. Other strategies proposed to overcome inadequate clopidogrel responsiveness with goal to reduce the risk of recurrent ischemic event include increasing clopidogrel doses, use of triple antiplatelet therapy, and switching to other P2Y12 antiplatelet agents.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


