Personalized Pulmonary Medicine
INTRODUCTION
Since the first draft sequences of the human genome were completed in 2001,1,2 medical research has increasingly focused on the utilization of genetic and genomic profiling in the prediction of disease susceptibility and natural history, as well as drug response and drug development. Personalized medicine can be defined as an approach to medicine in which medical decisions are tailored to the individual patient. In theory, personalized medicine will avoid costly and prolonged trial and error approaches resulting in unwanted therapeutic side effects or diminished treatment efficacy. Diagnostically, personalized medicine uses molecular tracking to signal risk of disease on a genetic level, which may identify disease presence before clinical indications and symptoms appear. Thus, personalized medicine enhances the focus on preventive medicine at the primary, secondary, and tertiary levels. Fully realized, personalized medicine has the potential to facilitate early diagnosis and/or prevention of disease and selection of optimal therapeutic choices with minimal attendant side effects for established disease states. The potential savings, from both a financial and quality-of-life perspective, are enormous.
Much of the efforts to adopt personalized medicine into clinical practice have centered on genetic approaches, as sequence changes in deoxyribonucleic acid (DNA) have been closely associated with a wide range of disease susceptibilities and therapeutic responses. However, the “omics” era includes enhanced focus on cellular and metabolic changes downstream of DNA sequence variation including genomics or transcriptomics (the analysis of gene expression), proteomics (the analysis of protein changes), and metabolomics (the analysis of end products of cellular metabolism). Adding to genomic complexity are the so-called epigenetic changes, the study of changes in gene expression or cellular phenotype caused by mechanisms other than changes in DNA sequence, such as DNA methylation, post-translational modification of gene expression, and microRNA (see further in Chapter 7). Each of these genomic study types has the potential to serve as a biomarker in a personalized medicine context.
In this chapter, the foundations of personalized pulmonary medicine will be reviewed and current approaches designed to facilitate a personalized approach to the diagnosis and treatment of pulmonary disorders will be discussed, including specific examples of personalized approaches currently being implemented in clinical practice. We provide overviews of human genetics, personalized pulmonary diagnostic testing, pharmacogenomics, biomarkers, and future implementation as they relate to personalized respiratory medicine.
DETERMINANTS OF THE CLINICAL UTILITY OF A GENETIC TEST
The predictive power of a genetic test is a function of four interdependent estimable parameters: (1) the heritability of the trait; (2) the penetrance of the tested genetic variant; (3) allelic heterogeneity; and (4) the variant allele frequency.
Heritability can be defined as the proportion of disease risk that is explained by genetic determinants. Heritability is highest (∼100%) among monogenic diseases like cystic fibrosis (CF), while common diseases such as asthma or obstructive sleep apnea have more modest estimates (30%–60%), with a substantial proportion of disease risk is due to environmental factors. The heritability for many of the commonly measured pulmonary traits is modest (Table 9-1).
TABLE 9-1 Heritability Estimates in Pulmonary Medicine
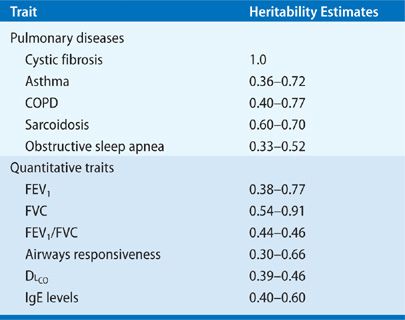
Penetrance is defined as the likelihood of the disease among individuals who carry the risk genotype (Fig. 9-1). Penetrance is highest for variants implicated in rare monogenic disorders, approaching 100% for the ΔF508 mutation in CF, and is lowest for more frequent genetic variants associated with common diseases. Wide ranges of intermediate penetrance have also been reported in pulmonary disease, from 90% for FLCN mutations in Birt–Hogg–Dubé syndrome (BHD)3 to 20% to 50% for BMPR2 mutations in familial pulmonary hypertension4 and the MUC5B promoter polymorphism in familial pulmonary fibrosis.5 Penetrance is phenotype-dependent. For example, in BHD, although ∼90% of patients with FLCN mutations have radiographic evidence of cystic lung disease, dermatologic findings are seen in only ∼60%, and spontaneous pneumothorax is observed in only 38% of patients.3 Similarly, though virtually all PiZ homozygotes have markedly reduced circulating levels of alpha 1-antitrypsin levels, only a subset of these individuals manifests symptomatic emphysema.6
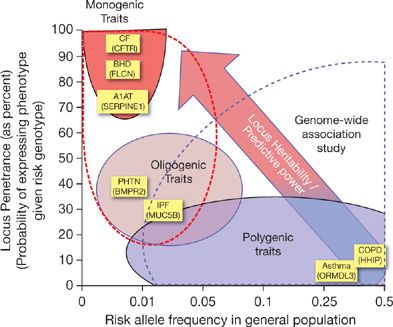
Figure 9-1 The relationship between allele frequency, locus heritability, predictive power, and genetic mapping strategies: Monogenic diseases, such as cystic fibrosis (CF), Birt–Hogg–Dubé syndrome (BHD), and alpha 1-antitrypsin deficiency (A1AT) are caused by highly penetrant, deleterious causal mutations that can be detected by parametric linkage analysis. Polygenic, complex traits, such as asthma and COPD, are caused by numerous genes (dozens to hundreds) harboring variants of much weaker genetic effect. Genome-wide association studies can identify the subset of these variants, but their individual predictive value is low. Oligogenic traits, like familial forms of pulmonary hypertension (PHTN) and idiopathic pulmonary fibrosis (IPF), are caused by variants in a handful of genes with both intermediate frequencies and intermediate penetrance functions.
Allelic heterogeneity refers to the existence of more than one causal allele present in the reference population. Though one variant often predominates as the most prevalent disease allele (e.g., the Z allele in 85% in alpha 1-antitrypsin deficiency), numerous pathogenic variants are typically described, including more than 20 SERPINE1 mutations in alpha 1-antitrypsin, 50 FLCN mutations in BHD, and more than 1800 pathogenic CFTR variants in CF. Locus heterogeneity is a situation where the same phenotype can manifest through mutations in different genes. For example, familial bronchiectasis resulting from primary ciliary dysfunction has been mapped to no fewer than 18 genes that code for distinct proteins of the ciliary apparatus. There are numerous reports of confirmed primary ciliary dyskinesia where only one of two pathogenic mutations is identified in any one gene,7,8 suggesting the potential for digenic inheritance, where the combination of two mutations in different genes is sufficient to cause disease.
The influence of variant allele frequency on the predictive power of a genetic test is complex, and depends largely on the strength of the genetic effect conferred (the locus heritability) and the disease prevalence. Rare monogenic lung diseases typically result from highly penetrant, low frequency (<1%) variants situated in highly conserved protein-coding sequences. Most common disease susceptibility variants reported are of high frequency (>5% in the general population) but individually are of weak effect. In many instances, the population risk allele frequency exceeds the population disease prevalence (such as the asthma-associated ORMDL3/GSDMB risk haplotype, frequency of 62% in asthmatics). In these cases, the specificity of the variant is quite weak. In contrast, rare variants of high penetrance implicated in common disease will have high specificity, yet their low population prevalence diminishes their clinical predictive value, as sensitivity and negative predictive values will be poor.
GENETIC TESTING
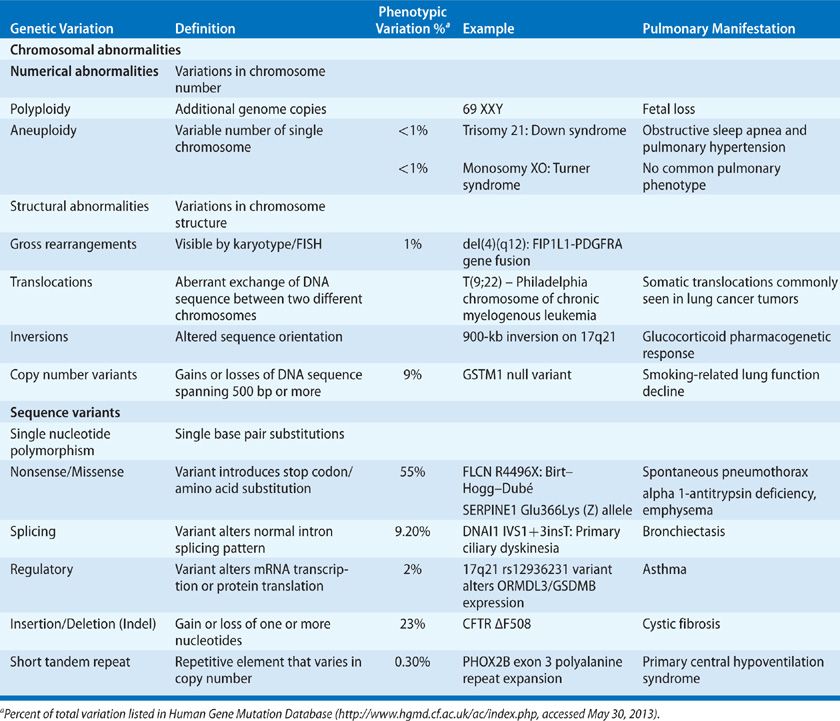
All of the major classes of genetic variation – including single nucleotide substitutions, insertions and deletions, copy number variants, and larger, more complex structural variants – have been described in pulmonary disease (Table 9-2), and no single technology has been developed that can reliably survey all these forms simultaneously, though next-generation sequencing (NGS) technologies are emerging as one possible solution. The rapid pace of technological advancement and the vast number of technologies in current use preclude comprehensive survey of all aspects of DNA technologies here. Instead, we emphasize NGS technology as a current and future direction for use in clinical practice.
TABLE 9-2 Types of Genetic Variation and their Relative Contribution to Disease Phenotype
Whole-genome sequencing. NGS platforms enable DNA sequencing in a highly paralleled fashion, without the need for predefined sequence-dependent hybridization. These methods generate short sequence reads (30–135 bases in length), sampled randomly from the target sample. With these methods, whole-genome sequencing of nearly all genomic regions is now feasible, and is being widely implemented for both research and clinical purposes. The massive parallel nature of these techniques, and their lack of reliance on Sanger chemistries, have resulted in dramatic reductions in cost (now $1500–$2000) for whole-genome sequence, favorably raising the prospects for comprehensive genomic testing and personalized medicine.
The major technical challenge, and primary determinant of cost, is achieving sufficient base-calling accuracy. Given the very large numbers of both the bases being called (billions) and polymorphisms per genome (∼3–4 million), accuracies of >99.9% are needed to limit the number of spurious findings (both false positive and false negative). Such accuracy can be achieved by ensuring high read depth—the number of times a given base is sequenced. For clinical purposes, minimum average read depths of 30 to 40 times provide reasonable accuracies. For whole genomes, such coverage can only be achieved by performing multiple sequencing runs, increasing costs considerably. More expensive are the downstream costs related to the involved analytical processes of quality assessment, sequence annotation, and variant classification (see below). Additional costs are incurred from the current clinical guidelines that mandate independent technical validation of all actionable (clinically relevant, reportable) variants by Sanger sequencing. Thus, while the technical cost continues to fall, current total charges (often exceeding $10,000 per clinical genome) preclude widespread adoption of whole genome sequencing at present.
Targeted sequencing panels. While use of whole-genome sequencing in clinical practice remains modest, there is increasing utility in highly focused, targeted NGS of selected genes or gene regions, which is achieved by “pulling-down” DNA segments of interest using oligonucleotide filters (i.e., probe sets that bind complementary sequence). These filters can be customized to target specific sequences, ranging from a handful of genes to all gene-coding regions (i.e., whole exome filters). This approach markedly reduces the size of targeted sequence length (e.g., an exome sequence is only 3% of the total genome sequence), with numerous advantages for clinical implementation. The marked reduction in target size accommodates clinically reliable read depths in one sequence run, reduces analytic costs (as there is less sequence to annotate), and reduces follow-up validation costs.
Pulmonary-specific sequencing panels. Targeted sequencing of selected genes implicated in pulmonary disease is available commercially through several companies and clinical laboratories. The services provided differ from each other primarily in the gene content of the tests, which are frequently organized in discrete gene panels. Most panels are designed to assess selected gene sets implicated in disorders typified by a particular clinical or radiographic manifestation (i.e., a pulmonary fibrosis panel) or a shared molecular defect (i.e., ciliopathy panel). The largest panels available are those for workup of pulmonary fibrosis and bronchiectasis. Due to technical limitations and the constant pace of novel disease-gene discovery, few panels can be considered fully comprehensive. Yet, most offer coverage of the most commonly implicated genes being considered clinically. Panels of 5 to 20 genes typically cost $2500 to $6000, in comparison to the cost of clinical resequencing of individual genes (on average $1500–$2000). Thus, when more than one gene is clinically suspected, or for diseases with known locus heterogeneity, diagnostic panels offer greater cost efficiency over single gene resequencing.
More broad sequencing panels may have particular value in the evaluation of patients with more complex presentations (e.g., patients presenting with a combination of bronchiectatic and fibrotic features, or with parenchymal lung disease disproportionate to the degree of concomitant pulmonary hypertension). When a genetic basis is suspected, massively paralleled sequencing across panels of genes may help narrow the differential diagnosis to one or two disorders. Currently, only one laboratory – the Laboratory for Molecular Medicine at the Partners HealthCare Center for Personalized Genetic Medicine – offers combination panel testing, at incremental cost over one panel. Though possibly more cost-effective over current diagnostic strategies, the value of such an approach to the workup of patients with complex presentations remains unclear.
We emphasized that, before ordering more expansive tests, patients must be made aware that surveys of larger panels of genes increase the likelihood of identifying variants of unknown clinical significance (so-called VUS). Patients must be counseled pretest regarding the uncertainty these findings may impart. Genetic test results classify variants according to their likelihood as disease-causing variants. Variants classified as “pathogenic” are those firmly implicated as disease causing due to their demonstrated functional impact, their localization to a highly conserved functional genomic sequence, their presence in affected individuals only, and their strong segregation with disease. So-called “likely pathogenic” variants have many, but not all, of these features. “Benign” variants are those that are common among healthy controls, have no demonstrable function, and do not segregate with disease in families. Counseling of patients regarding these three classes of variants is often unambiguous. In contrast, VUS variants fall into a grey zone, in that they do have features that suggest functional impact but are either observed in unaffected subjects or segregate imperfectly in affected pedigrees (features that suggest incomplete penetrance). Patients must understand the possibility of such findings, and their implications on subsequent workup and management.
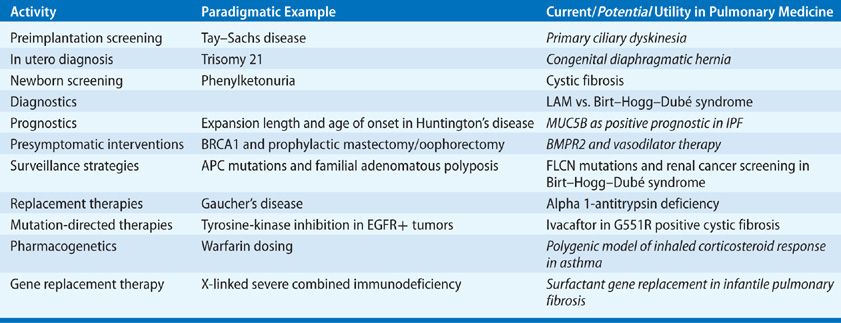
Clinical indications for genetic testing. Table 9-3 lists the most common uses of genetic information in clinical practice, both for the patient and at-risk family members. Diagnostic genetic testing is of greatest value when knowledge of the specific gene or mutation will directly impact clinical management. In these cases, genetic testing should be offered to patients in whom the possibility of defining an actionable variant is high, including those with specific disease presentations compatible with the diagnosis of interest, at-risk family members of patients with confirmed molecular diagnoses, and subjects with strong family histories. Some examples are as follows:
• Confirmation of alpha 1-antitrypsin deficiency in patients with emphysema and lung function decline identifies a small subset of COPD patients who may benefit from replacement therapy.
• Identification of Class III CFTR genotypes (particularly the G551R variant) in patients with CF. These patients are candidates for mutation-specific CFTR potentiating therapy with ivacaftor.9
• Differentiating molecular forms of familial idiopathic pulmonary fibrosis: (1) Patients with SFTPC mutations may benefit from hydroxychloroquine treatment10; (2) TERC and TERT mutations help identify patients with short telomere syndrome (STS) at risk of marrow and liver failure, including during the post-lung transplant period; (3) Hermansky–Pudlak syndrome (HPS) can often be overlooked clinically due to subtle neurologic (nystagmus) and dermatologic (albinism) features, though these patients are at risk for a potentially severe, but treatable (with DDAVP), bleeding diathesis.
TABLE 9-3 Utility of Genetic Information in Clinical Practice
For highly penetrant monogenic diseases, the implications of a positive test result are often profound, even in instances where gene- or mutation-specific therapies are not yet available. Examples include preclinical identification of patients at risk for malignancy (e.g., in BHD or LAM), accelerated lung function decline (in LAM, familial fibrosis), or pulmonary hypertension, who can be more closely followed enabling earlier intervention, and who can be more vigorously counseled regarding the benefits of tobacco smoke avoidance. An illustrative example is provided in Figure 9-2, depicting the value of testing in a family pedigree with BHD. For autosomal dominant diseases of variable penetrance, and for recessive disease, genetic testing offers the ability to identify at-risk carriers prior to the onset of symptoms, providing opportunities for early diagnosis. Negative test results are also of value, providing reassurance to relatives that they are not at risk of developing an illness they may have witnessed afflicting their relatives.
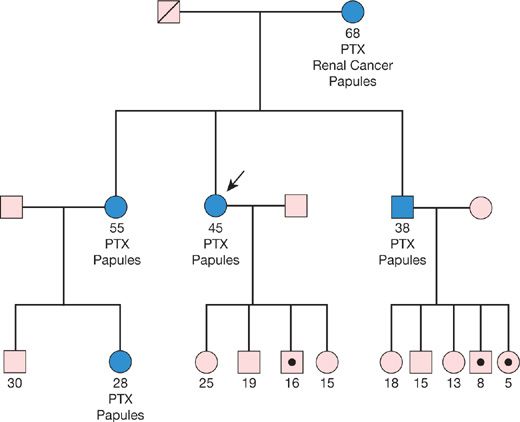
Figure 9-2 Birt–Hogg–Dubé (BHD) syndrome: Index patient (arrow) confirmed to harbor a pathogenic FLCN gene mutation. The maternal history of renal cancer places other mutation carriers in this family at risk of both renal and other malignancies. In this pedigree, the large number of reportedly unaffected offspring of two carriers is unusual, given a 50% probability of transmission from carrier to offspring. More likely, the mutation was passed on (hypothetically denoted by dot), but these carriers have not yet developed clinical manifestations, possibly due to their younger age. Such individuals would be at risk of eventually developing BHD, including malignancy. Therefore, confirmation of carrier status among at-risk, but seemingly unaffected, family members is warranted.
Genetic counseling in pulmonary medicine. The inherently predictive, personal, and irreversible nature of an individual’s genetic code distinguishes genetic from other forms of clinical testing. The psychological impact of genetic test results, whether positive or negative, cannot be underestimated. Feelings of inadequacy or imperfection, a sense of inevitable doom and therapeutic nihilism due to a genetic “fate” are not uncommon with a confirmed genetic diagnosis. Conversely, negative results can lead to a false sense of invincibility, leading to unhealthy behaviors (e.g., continuing to smoke). Inherently, the potential impact of genetic testing extends beyond the patient, implicating all blood relatives. Moreover, test results can impact family dynamics and the way in which individual family members react to their test results. For example, though most at-risk family members will be relieved by a negative test result, some experience a deep sense of guilt (so-called “survivors guilt”) in relation to their affected family members. Finally, despite legislation at state and federal levels protecting patients from genetic discrimination in the workplace and in access to health care insurance, patients must be informed regarding this risk in other contexts (e.g., the impact on eligibility for life insurance policies).
For all these reasons, it is advised that genetic testing be offered only by, or in consultation with, experienced providers familiar with these issues, including certified genetic counselors, who can adequately address the medical, psychological, and familial implications of test results. We recommend initiating these discussions before testing, so that patients are empowered to provide truly informed consent. Patients should be made aware of the medical implications of both positive and negative results, and should be advised regarding the impact these results may have on them and their family members. Genetic pretest counseling should also address the potential identification of variants of unclear significance.
MUTATION-SPECIFIC THERAPIES IN MONOGENIC LUNG DISEASE
One of the primary goals of genetic classification of disease is the development of mutation-specific therapies. Such activities have traditionally focused on the development of replacement therapies for loss-of-function recessive diseases (i.e., recombinant human alpha 1-proteinase inhibitor replacement therapies). Newer pharmacologic approaches, including small molecule screens and others informed by the functional impact of specific mutations are emerging.
 TYROSINE-KINASE INHIBITION FOR HYPEREOSINOPHILIC SYNDROME
TYROSINE-KINASE INHIBITION FOR HYPEREOSINOPHILIC SYNDROME
Imatinib was identified as a therapy for chronic myelogenous leukemia (CML). However, once the functional mechanisms underlying its therapeutic effects were elucidated, other therapeutic uses were identified. The treatment of hypereosinophilic syndrome (HES) is one such example. Subsets of HES patients harbor a chromosome 4q interstitial deletion that results in the creation of a fusion of the FIP1L1 and PDGFRA genes. The resultant fusion protein has tyrosine-kinase activity similar to that of BCR-ABL, the target site in CML and HES patients positive for the FIP1L1-PDGFRA rearrangement respond positively to imatinib therapy.11,12 FIP1L1-PDGFRA-negative HES patients do not. Similar to CML, mutations of the tyrosine-kinase binding site have been reported upon HES relapse.
 MUTATION-SPECIFIC THERAPIES FOR CYSTIC FIBROSIS
MUTATION-SPECIFIC THERAPIES FOR CYSTIC FIBROSIS
In 2011, positive results of a randomized clinical trial of a novel therapy for G551D-positive CF were reported (Fig. 9-3).9 In a 48-week, randomized, double-blind, placebo-controlled trial, 167 patients received either placebo or ivacaftor—an oral agent that “potentiates” CFTR activity by prolonging channel opening times and augmenting transmembrane chloride transportation. Compared to patients receiving placebo, patients treated with ivacaftor demonstrated significant improvements in FEV1, fewer respiratory exacerbations, improved quality of life, and a positive weight gain. The ∼50% reduction in sweat chloride levels to those approaching those observed in asymptomatic CFTR mutation carriers (i.e., heterozygotes) serves as an elegant molecular–clinical correlate, suggesting selective effect on the G551D+ channel, but not ΔF508+ channels. While G551D and other variants likely to benefit via ivacaftor are observed in only ∼6% of patients, their identification opens new therapeutic options for this subset of CF patients. The advent of this novel treatment strategy provides motivation for the development of additional agents that target other CFTR mutation classes (including the most common CFTR mutation—ΔF508), and the application of similar strategies for the treatment of other genetic diseases.
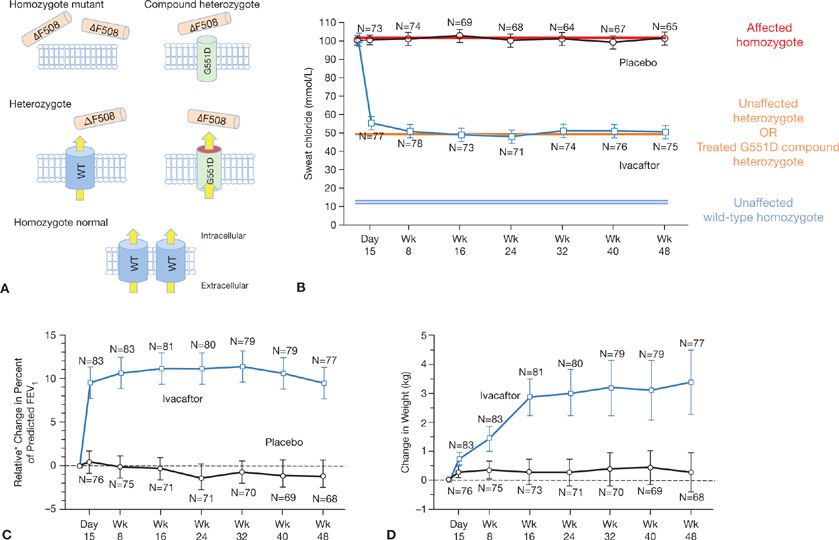
Figure 9-3 Mutation-specific therapeutic action of ivacaftor in cystic fibrosis: A, B. The additive effect of CFTR mutations on epithelial chloride efflux and sweat chloride levels, and the effect of ivacaftor. Individuals with two normal alleles have normal chloride efflux (yellow arrows), and a corresponding normal sweat chloride test (blue double line). Individuals with two mutated alleles demonstrate markedly reduced chloride efflux and elevated sweat chloride (red line). Heterozygotes with ∼50% normal functional chloride channels on epithelial cell surfaces manifest intermediate reductions in both chloride efflux and sweat chloride responses (orange line). G551D compound heterozygotes treated with ivacaftor (red disc) demonstrate sweat chloride levels similar to heterozygote carriers. C, D. Therapeutic efficacy of ivacaftor: Patients randomized to ivacaftor demonstrated improvements in relative change in percent of predicted FEV1 (C) and weight (D) over 48 weeks, in addition to improvements in symptoms and in quality of life scores (not shown). *Original report erroneously labeled figure as Absolute (not relative) change in percent of predicted FEV1. (B–D. Reproduced with permission from Ramsey BW, Davies J, McElvaney NG, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365(18):1663–1672.)
BIOMARKERS IN PULMONARY MEDICINE
Biomarkers have been defined as “biological characteristics that can be objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes, or pharmacological responses to a therapeutic intervention”.13 Ideal biomarker characteristics have been espoused (Table 9-4).14 There are two major types of biomarkers applicable to clinical medicine: (1) biomarkers of exposure, which are used in risk prediction; and (2) biomarkers of disease, which are used in the screening, diagnosis, and monitoring of disease progression, as well as response to therapy. Thus, biomarkers have the potential to support clinical decisions, from diagnosis to treatment planning; to improve tailored treatment strategies; to avoid over- or undertreatment and adverse side effects; and to enhance prognosis and cost-effectiveness.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


