Pediatric Heart Transplantation
Charles B. Huddleston
The first pediatric heart transplant was performed by Dr. Adrian Kantrowitz on a small infant with tricuspid atresia in 1967. The child died soon after the procedure but this ushered in the concept of heart replacement for unreconstructable congenital heart disease. There were very few transplants in children from the late 1960s into the early 1980s. From the mid-1980s onward there was a steady increase in the number of pediatric transplants related in large part to the development of cyclosporine as an immunosuppressant as well as the successes of the group at Loma Linda University led by Dr. Leonard Bailey in children and that of Norman Shumway at Stanford University. Approximately 400 to 450 heart transplants are performed in children per year throughout the world. This number has been relatively static over the past 15 years. Survival in children is similar to that seen in adults at 5 to 10 years post-transplant but improves at 15 years (48% vs. 34%). A number of changes have occurred in pediatric heart transplantation over the past 20 years. Transplantation as primary therapy for any congenital heart disease (hypoplastic left heart syndrome in particular) is unusual. There has been a steady increase in the use of ventricular assist for children with end-stage heart failure, usually due to cardiomyopathy. This chapter will reflect some of these changes. Although “pediatric heart transplantation” should be limited by age, I will also include transplantation in adults with congenital diagnoses.
 INDICATIONS
INDICATIONSHeart transplantation in children has two basic indications—cardiomyopathy and congenital heart disease. On balance, these indications are approximately evenly split, although the congenital diagnosis predominates in younger patients and cardiomyopathy accounts for the majority of patients in the teenage group. The technical aspects of the procedure in cardiomyopathy are no different than that used in adults. The technical challenges lie in replacing the heart in a child with disordered anatomic arrangements of the great vessels (arteries and veins) and with anatomic derangements made so by prior corrective or palliative procedures. Hypoplastic left heart syndrome was a major indication for heart transplantation in the early 1990s in large part due to the poor survival following the Norwood procedure. Current survival with reconstructive first-stage palliation has been approximately 80% for the participants in the congenital registry of the Society of Thoracic Surgeons. In addition to this, transplant waiting time mortality may be as much as 20% to 25% in this group of neonates. It is impractical to consider transplantation for even half of infants with hypoplastic left heart syndrome in that at least 500 infants are born each year with this congenital lesion in the United States where less than 100 donors in this age group are available. Among those with congenital heart disease as the primary diagnosis, most have single-ventricle anatomy and may be at various stages of the reconstructive pathway from diagnosis to Fontan. Whereas a better understanding of the implications of early palliation for these patients has led to more successful outcomes with the Fontan procedure, the survival curves for those patients clearly declines at a much faster pace than the normal population or even the overall survival curve for children with congenital heart disease. These patients present with need for special consideration as far as the technical aspects of the transplant, the presence of significant elevation in pulmonary vascular resistance, the presence of preformed antibodies, and the presence of multiple small aortopulmonary collaterals. There remain a significant number of young adults who survived atrial switch procedures for transposition of the great arteries. Many (all?) of those patients will ultimately develop heart failure. This group will be out of the age range for pediatrics, but a clear understanding of the anatomy involved is necessary for a technically successful outcome.
CONTRAINDICATIONS
Absolute contraindications to heart transplantation include ongoing malignancy, multisystem organ failure refractory to intensive treatment for heart failure, uncontrolled infection, and significant psychosocial issues. The presence of renal or hepatic dysfunction depends in large part on the degree and whether the patient might be candidate for combined or staged transplantation of either the liver or kidney. Elevated bilirubin has consistently been a marker for poor outcomes in heart transplantation. Renal dysfunction may be particularly problematic as both cyclosporine and tacrolimus (the primary drugs of immunosuppressant regimens posttransplant) have significant nephrotoxic side effects. The availability of effective mechanical support in children has broadened the recipient pool to some degree as those patients with significant organ dysfunction can be stabilized with ventricular assist devices and made better candidates.
Elevated pulmonary vascular resistance has been a classic risk factor for heart transplantation. The availability of multiple drugs to treat this in both the acute and chronic setting has altered the approach to this. As a baseline, the pulmonary vascular resistance should be <6 to 8 Woods units and the transpulmonary gradient should be <15 mmHg. It is often difficult to obtain accurate values of one or both of these because of congenital or acquired anatomy, resulting in multiple sources of pulmonary blood flow and difficulty accessing the pulmonary vasculature at the time of the cardiac catheterization. Patients deemed to be at high risk due to elevated pulmonary vascular resistance should be extensively studied to evaluate their response to a variety of pulmonary vasodilators. A lack of response acutely should be followed by long-term treatment with pulmonary vasodilators such as bosentan or prostacyclin in combination with inotropic support and then re-studied. Occasionally, an open lung biopsy may be of use to evaluate the presence of coexisting pulmonary parenchymal
disease. Patients with a very high systemic ventricular end-diastolic pressure may benefit from ventricular assist device implantation for several weeks and then reevaluated. Failing these measures, heart-lung transplantation would be an alternative, recognizing that the long-term prognosis following this transplant procedure is significantly worse than isolated heart transplantation.
disease. Patients with a very high systemic ventricular end-diastolic pressure may benefit from ventricular assist device implantation for several weeks and then reevaluated. Failing these measures, heart-lung transplantation would be an alternative, recognizing that the long-term prognosis following this transplant procedure is significantly worse than isolated heart transplantation.
DONOR ASSESSMENT/MANAGEMENT
After the usual criteria for donor acceptance for organ acceptance have been met (blood-type compatibility, absence of transmissible disease), size match and organ function are then considered. Size match is of particular concern for small infants as very small donors are unusual. Accepting a heart from a donor three times the weight of the recipient will generally work out well, realizing that it may be necessary to open the left pleural space, remove some pericardium, and leave the sternum open for a few days posttransplant. For older children and teenagers, the range is usually 20% above and below the recipient weight. Many surgeons are of the opinion that a larger donor is preferred for patients with borderline elevated pulmonary vascular resistance; there are little data to support that position. Blood-type compatibility has been challenged in infants up to 1 year of age with results that mimic earlier results, albeit with more a complex immunosuppressive regimen.
The assessment of donor heart function is typically done with echocardiography only. The donor should be on only a modest degree of inotropic support with satisfactory blood pressure and evidence of good cardiac output clinically. Evaluation of the cardiac markers of ischemia (myocardial fraction of creatine phosphokinase and cardiac troponin I) should be routine. Donor hearts with borderline systolic function may be resuscitated using intravenous infusion of triiodothyronine. The basis of this is evidence that brain death is associated with reduction in cortisol and thyroid hormone production. Vasopressin is often necessary in the treatment of diabetes insipidus; the use of this drug will often allow a reduction in inotropic support. There are no absolute guidelines for the upper limit of inotropic support allowable for a donor heart to avoid posttransplant primary graft dysfunction, but generally one should avoid those requiring high doses of two or more.
POST-TRANSPLANT COMPLICATIONS
Graft Failure
This is the most common cause of early posttransplant deaths, especially in those transplanted at <1 year of age. Graft failure may be related to poor preservation, poor donor selection, early rejection, pulmonary hypertension, or technical issues. Support for graft failure posttransplant is usually ECMO. Intra-aortic balloon counterpulsation is generally ineffective in children with fast heart rates and a compliant aorta. Ventricular assist is feasible with current small devices available. Pulmonary hypertension as a cause of early graft failure usually presents with elevated central venous pressure, tricuspid valve regurgitation, and low cardiac output. The pulmonary artery and right ventricular pressure may not be elevated because the donor right ventricle may not be able to generate high pressures. The usual measures of treatment would be sedation with neuromuscular paralysis, inhaled nitric oxide, and inotropic support. Occasionally, mechanical support is necessary. One must be certain that there are no technical problems with the pulmonary artery anatomy or anastomosis, particularly in the setting of prior palliative operations involving the pulmonary arteries.
Rejection
The incidence of acute rejection is lower in infants than in older children and teenagers. Nonetheless, surveillance is necessary in all age groups. A high index of suspicion should be maintained in patients with preformed antibodies going into the transplant procedure. The diagnosis of cell-mediated rejection is relatively straightforward using endomyocardial biopsy material. Antibody-mediated rejection, however, is significantly more difficult to diagnose. Although biopsy material can be stained for complement factors (Cd4 being the most common), this is not completely reliable. Evidence of poor cardiac function clinically and by echocardiography with a biopsy that is negative for cellular rejection should prompt this diagnosis. Treatment for acute cellular rejection is high-dose steroids (10 mg/kg methylprednisolone intravenous) daily for 3 days. Antibody-mediated rejection requires plasmapheresis as well as drugs aimed at reducing the production of antibodies.
Bleeding
These patients often come to transplant having had multiple prior operations with the anticipated adhesions. In addition, they may be taking anticoagulants or have disordered hemostatic mechanism due to long-standing heart failure. Additional use of blood products is anticipated.
Other Complications
Infection is a constant risk for these patients due to the immunosuppression necessary. Prophylaxis against Pneumocystis jirovecii is necessary using either sulfamethoxazole/trimethoprim daily or inhaled pentamidine monthly. Renal insufficiency related to marginal pretransplant renal function and calcineurin inhibitors posttransplant is relatively common to some degree and may occasionally require temporary dialysis. Seizures may occur in 5% to 10% of patients due to posterior reversible encephalopathy syndrome. Posttransplant coronary vasculopathy is a long-term complication, occurring in 35% of patients by 10 years posttransplant; this is much more common in adults, present in more than 50% at 10 years posttransplant. Lymphoproliferative disorder and other malignancies are present in approximately 10% of patients in long-term follow-up.
OPERATIVE TECHNIQUES
General Comments
As mentioned above, the technique of transplantation for patients with cardiomyopathy is the same as with adult transplantation and will not be presented further in this chapter. The focus will be on issues that are unique to congenital heart disease. The number of different combinations of congenital anomalies and their anatomic nuances preclude an encyclopedic description of each method of recipient preparation and donor implant. The principles presented for the conditions described can be adapted for each individual situation. The patients with single-ventricle anomalies provide the greatest challenges primarily because of the abnormalities in situs and venous anatomy as well as the obligatory pulmonary artery anomalies associated with the palliative procedures that these children have undergone in the past. Careful planning of the procedure by reviewing prior operative notes, cardiac catheterizations, and other imaging studies is crucial to conducting a safe operation. A computed tomography (CT) scan with contrast is particularly
helpful in providing landmarks for careful sternal re-entry. It is important to obtain sufficient donor tissue for whatever reconstruction is necessary, usually the superior vena cava and branch pulmonary arteries. In the setting of multiorgan retrieval where lungs in particular are being retrieved, additional length of aorta should be obtained to use if pulmonary artery reconstruction is anticipated. Alternatively, some of the native tissue that would otherwise be discarded with the recipient heart may be suitable for patches. Caval anastomoses (as opposed to an atrial anastomosis) have become the standard for transplantation with the exception of small infants where the risk of anastomotic narrowing of the superior vena cava is relatively high.
helpful in providing landmarks for careful sternal re-entry. It is important to obtain sufficient donor tissue for whatever reconstruction is necessary, usually the superior vena cava and branch pulmonary arteries. In the setting of multiorgan retrieval where lungs in particular are being retrieved, additional length of aorta should be obtained to use if pulmonary artery reconstruction is anticipated. Alternatively, some of the native tissue that would otherwise be discarded with the recipient heart may be suitable for patches. Caval anastomoses (as opposed to an atrial anastomosis) have become the standard for transplantation with the exception of small infants where the risk of anastomotic narrowing of the superior vena cava is relatively high.
Hypoplastic Left Heart Syndrome
Although reconstructive surgery has generally supplanted transplantation as primary therapy for HLHS, there are circumstances where the risks of the Norwood procedure are prohibitive and the balance shifts toward transplantation. These risk factors are pulmonary valve stenosis or regurgitation, severe right ventricular dysfunction, severe tricuspid valve regurgitation, or syndromic infants (e.g., Turner syndrome). Small size (<2.5 kg) is another risk factor for the Norwood procedure, but finding a suitably small donor is problematic.
Donor Procurement
The major concern is to acquire all of the aortic arch and the proximal descending thoracic aorta to the level of the ligamentum arteriosum. This provides sufficient donor aorta for the arch reconstruction. At the time of donor operation, the innominate vein may be ligated to get better access to the arch vessels. After aortic clamping and cardioplegia administration, the innominate, left carotid, and left subclavian arteries are divided at their origins. The descending aorta is taken just beyond the ligamentum arteriosum. The rest of the procurement proceeds as per the usual technique.
Recipient Operation
The ductus arteriosus, branch pulmonary arteries, and aortic arch with its branches are dissected out extensively. When dissecting out the ductus arteriosus and during the distal arch reconstruction, care must be taken to avoid injury to the recurrent laryngeal nerve. Cannulation for arterial inflow may be performed by one of the three methods: via the main pulmonary artery with control of the branch pulmonary arteries, directly into the ductus arteriosus with ligation of the pulmonary artery end of the ductus, or via the innominate artery usually through a small graft sewn to the innominate. The last method provides the greatest flexibility for cannulation during the organ implant and potentially allows for regional perfusion during arch reconstruction so that no period of circulatory arrest is necessary. Bicaval cannulation is generally preferred and the patient is cooled to 18°C. While the cooling is proceeding, the donor heart is prepared by removing the cephalad aspect of the aortic arch leaving a “tongue” of aorta to reconstruct the arch (Fig. 99.1A). The recipient heart is excised, ligating the ascending aorta with a silk tie (Fig. 99.1B). The left atrial anastomosis is performed first. I prefer to perform
a portion of the right atrial anastomosis now because visualization will compromise coronary sinus blood return once the aortic reconstruction is completed and the clamp removed. At this point, either circulatory arrest or regional cerebral perfusion is initiated. Regional perfusion requires occlusion of the innominate artery proximal to the inflow graft sewn there and occlusion of the other arch branches as well as clamping the descending thoracic aorta 1 to 2 cm beyond the insertion of the ductus arteriosus. All the ductal tissues are excised and an incision is carried down into the aorta beyond the distal extent of ductal tissue. The remainder of the aortic arch is opened all the way to the distal ascending aorta (Fig. 99.1C). The aortic anastomosis is performed beginning distally and bringing the suture line all the way around the entire aortic arch. Following completion of this, the pulmonary artery anastomosis is performed in an end-to-end manner; the recipient MPA is often much larger than the donor. Finally, the transplant is completed with the anterior portion of the right atrial anastomosis (Fig. 99.1D).
a portion of the right atrial anastomosis now because visualization will compromise coronary sinus blood return once the aortic reconstruction is completed and the clamp removed. At this point, either circulatory arrest or regional cerebral perfusion is initiated. Regional perfusion requires occlusion of the innominate artery proximal to the inflow graft sewn there and occlusion of the other arch branches as well as clamping the descending thoracic aorta 1 to 2 cm beyond the insertion of the ductus arteriosus. All the ductal tissues are excised and an incision is carried down into the aorta beyond the distal extent of ductal tissue. The remainder of the aortic arch is opened all the way to the distal ascending aorta (Fig. 99.1C). The aortic anastomosis is performed beginning distally and bringing the suture line all the way around the entire aortic arch. Following completion of this, the pulmonary artery anastomosis is performed in an end-to-end manner; the recipient MPA is often much larger than the donor. Finally, the transplant is completed with the anterior portion of the right atrial anastomosis (Fig. 99.1D).
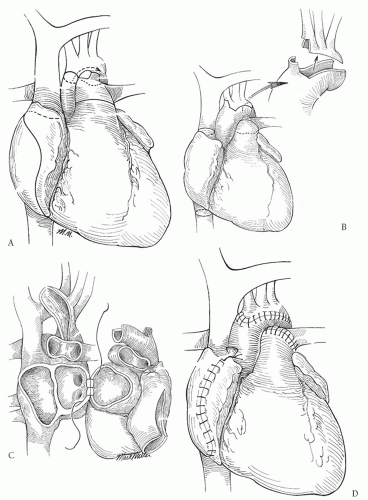 Fig. 99.1. Transplantation for hypoplastic left heart syndrome. (A) The recipient heart is removed leaving right and left atrial cuffs and dividing the main pulmonary artery between the bifurcation and the sinotubular ridge of the pulmonary valve. The ascending aorta is transected below the innominate artery and the underside of the aorta is opened beyond the insertion of the ductus arteriosus. (B) The donor heart should be retrieved with a segment of aortic arch all the way to the ligamentum arteriosus of the donor. The arch branches are removed leaving a “tongue” of aortic tissue for the arch reconstruction. (C) The transplant is performed with the left atrial anastomosis first, followed by the posterior right atrial anastomosis and then the arch reconstruction. (D) The pulmonary artery and anterior right atrial anastomosis are completed with the cross-clamp removed. |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


