An unconventional but effective starting point for an organization of pediatric epilepsies is the interictal EEG. As shown in Table 10.1, the various patterns encountered in clinical practice may be reduced to five discrete interictal EEG groups. There are two major domains: the organization of the background and the characteristics of the epileptiform activity, most importantly the morphology of the interictal epileptiform discharges (IEDs). By considering the age of onset, one can narrow down the epilepsy syndromes to two or three possibilities, and the predominant seizure type will easily guide one to the final diagnosis. In a minority, most particularly the familial epilepsies, verification of other similarly affected family members is critical for diagnosis. These groupings provide some powerful information: if there are genetic predispositions, they predict the mode of inheritance, they inform about the general prognosis, they have strong treatment implications, and they could be used as the basis for referral to tertiary epilepsy centers.
Organization of the Epilepsies by EEG Characteristics
Of course, this is the opposite of the way we as clinicians normally conduct our evaluations. A precise history and physical examination should always come first. It is the sine qua non portion of the epilepsy evaluation, the basis of all we do, and the most important daily activity of the child neurologist. A thorough history elicited with minimal interruption accompanied by close observation of the child and family allows us to make the interpersonal connections that are critical for trust and healing.
The interictal EEG, however, is a remarkably powerful tool and even though it sits in second place to our clinical assessment, it nevertheless informs us about the underlying pathophysiology in a manner that our naked senses never could. For many years, our predecessors have appreciated that the various epilepsy syndromes have different relative contributions of genetic and structural components. These thoughts were codified in the 2010 publication of the International League Against Epilepsy’s Classification Committee (1). In this chapter, we will see how the interictal EEG informs us about these fundamental characteristics. The premise is simple: genetic and structural factors have markedly different EEG signatures, which allows the EEG to effectively categorize the epilepsies. Indeed, the EEG findings can be used as an endophenotype to explore the genetic basis of susceptibility to epilepsy (2). This type of epilepsy syndrome organization is highly practical and simultaneously it reveals some fundamental principles about the causes of the epilepsies. Another remarkable fact is that this can be usually accomplished with a relatively brief sample of the awake and sleep interictal EEG.
THE FAMILIAL EPILEPSIES (EPILEPSIES WITH FREQUENTLY NORMAL INTERICTAL BACKGROUNDS)
One of the five broad categories of EEG features seen in children with epilepsy is a normal tracing. The precise percentage of normal EEGs seen in patients with epilepsy is difficult to determine, but it may be as low as 8% (3). One reason for a repeatedly normal tracing may be a remote location of an epileptogenic lesion—one that does not readily allow for detection using conventional scalp recording electrodes. Normal tracings are also seen in certain distinctive epilepsy syndromes that share a common characteristic: they are familial epilepsies that are inherited in an autosomal dominant fashion. These may be considered the best examples of familial epilepsies. Why these epilepsies most often present with normal interictal backgrounds is not entirely clear, but certainly, the normal background rhythms speak to the absence of cognitive impairment and disability in the vast majority of individuals with these epilepsies. Here we encounter the first EEG-epilepsy paradox: even though these epilepsies are strongly genetically determined and spikes in other conditions have a strong genetic component (vide infra), the most conspicuous feature of the background is the lack of interictal epileptiform activity in these familial epilepsies.
There are familial epilepsies for every epoch of pediatric life starting with the neonatal period, and continuing to infancy, childhood, and adolescence. These include benign familial neonatal epilepsy (BFNE), benign familial infantile epilepsy (BFIE), autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE), autosomal dominant epilepsy with auditory features (ADEAF), and other autosomal dominant temporal lobe epilepsies. It is obvious from the titles of these epilepsies that they vary in age of presentation, the brain region commonly involved, and the clinical manifestations. Generally, the combination of the clinical features and family history, along with the mostly normal interictal EEG data, is sufficient to make a diagnosis, but if confirmation is required, genetic testing can help. By and large, the outcome is favorable, although there are published cases of severe phenotypes (4).
Benign Familial Neonatal Epilepsy
BFNE usually begins on the second or third day of life and resolves within the first few months. During seizures, neonates may have tonic posturing, automatisms, apnea, or focal or generalized clonic activity. The seizures may rarely persist into adulthood. Typically, the interictal EEG is normal, though it may have focal or multifocal sharp waves, an excessively discontinuous tracing for age or the theta-pointu alternant pattern. The latter is characterized by rhythmic 4- to 7-Hz activity, with admixed sharp waves, with shifting laterality across the hemispheres, but it is by no means specific for this disorder. Authors describe a clinical sequence starting with hypertonia and followed by apnea, autonomic signs, facial movements, and limb clonus (5). In some of the recorded seizures, an electrodecrement heralds the event and may last up to 19 seconds, followed by 1- to 2-minute bilateral spikes or sharp waves (6), but focal seizures (7) and focal seizures with secondary spread have also been recorded (8). The disorder has been associated with mutations in the KCNQ2 and KCNQ3 voltage-gated potassium channels, responsible for the M-current (9). This slowly activating current regulates subthreshold neuronal excitability and therefore raises the possibility that a medication like retigabine may be helpful since it blocks the M-current. A closely related syndrome of benign neonatal-infantile seizures has been associated with a missense mutation in the SCN2A gene, which encodes the alpha-2 subunit of the voltage-gated sodium channel (2q24) (10).
Benign Familial Infantile Epilepsy
BFIE and sporadic forms (which may be similar disorders) have been reported by several different investigators working in different regions of the world (11–14). Affected infants have focal seizures, which may present with subtle behavioral arrest or, on the other extreme, apparent bilateral convulsive features. Eye version, staring, oral automatism, and oxygen desaturation are other common elements, and these are commensurate with the location of onset of the seizures. The interictal EEG is usually normal, though some BFIEs may have peculiar low- or medium-voltage vertex spikes or sharp waves followed by a slow wave with a dome-like morphology (12,15). Ictal discharges often arise from the temporal region or posterior quadrant. This finding is relatively nonspecific, since many focal seizures in infants arise from the same region. These disorders are inherited in an autosomal dominant fashion and several genes have been discovered, which often involve ion channels, but importantly, sometimes do not (16). Defects in the ATP1A2 gene may cause familial hemiplegic migraine and associated infantile seizures (17). When there is associated paroxysmal kinesigenic dyskinesia, mutations in the proline-rich transmembrane protein (PRPT2) have been reported (18). Similar mutations have been identified in Japanese children (19) and even, rarely, have been found in sporadic cases (20). The fact that only 6 of the 16 probands in the Japanese study tested positive indicates that there are clearly other genetic causes of BFIE still to be discovered.
Autosomal Dominant Nocturnal Frontal Lobe Epilepsy
Patients with ADNFLE have sudden awakenings with dystonic movements (tonic posturing) or violent movements. Since the EEG findings are usually relatively bland, the seizures may be mistaken for sleep disorders, nocturnal paroxysmal dystonia, or nonepileptic seizures. Rarely, the interictal EEG may show focal features such as slowing and spikes (21). Ictal recordings are more distinctive and are characterized by bifrontal beta activity or other rhythmic discharges (22) (Fig. 10.1). The ictal discharges may lateralize or even localize to a region, suggesting a focal structural lesion, and therefore, a careful family history is warranted before considering focal respective surgery in patients with nocturnal frontal lobe seizures. Genetic screening for the associated nicotinic acetylcholine receptor gene mutation (CHRNA4 and CHRNB2) is commercially available.
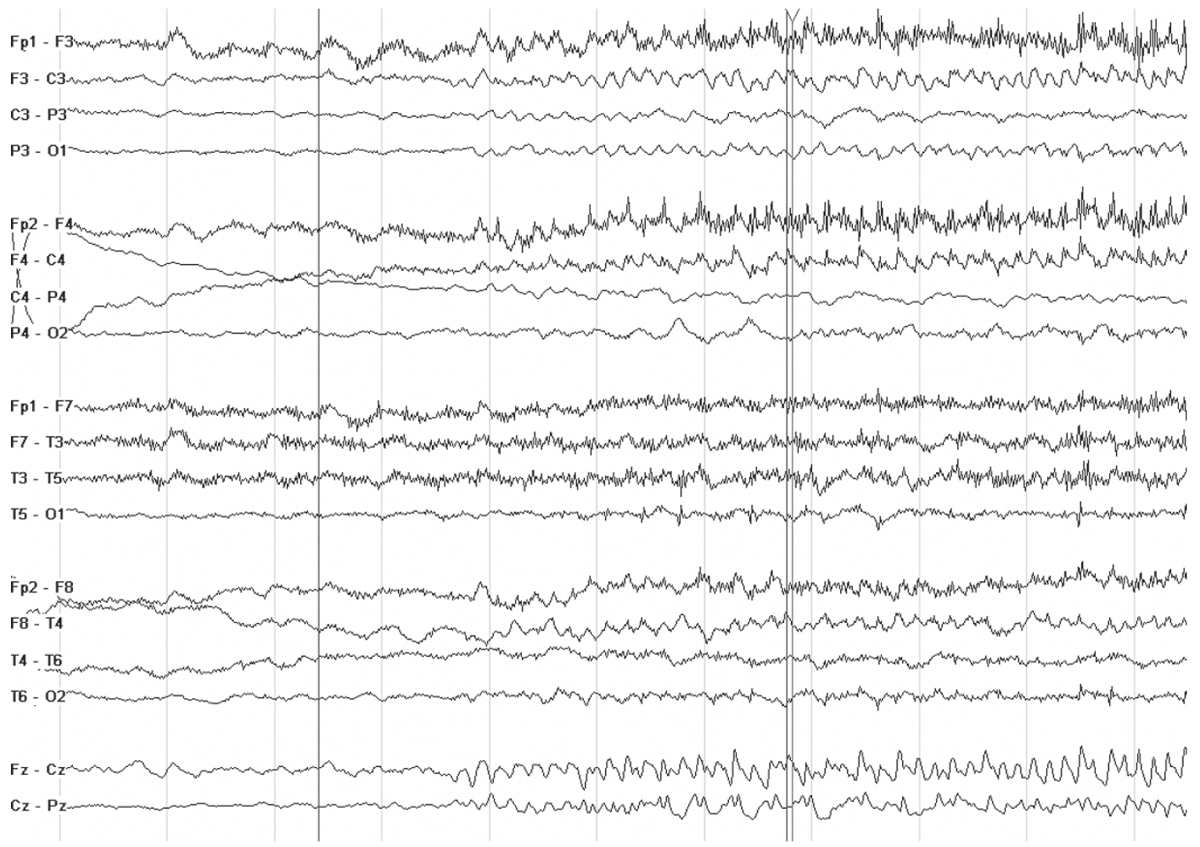
Figure 10.1: Ictal recording in a 10 year old boy with ADNFLE. Note the sudden change in the background as he awakens from sleep. There is some admixed low-voltage rhythmic fast activity in the frontal regions.
Familial Lateral Temporal Lobe Epilepsy or Autosomal Dominant Epilepsy with Auditory Features
Ottman described a pedigree in 1995 where most affected members reported seizures with auditory symptoms and named the syndrome autosomal dominant partial epilepsy with auditory features (ADPEAF [OMIM 600512]) (23). Subsequently, it was found that mutations of the leucine-rich glioma-inactivated 1 LGI1 gene caused this disorder (24). The onset of symptoms is usually in adolescence of early adult life, but childhood onset is also possible. It is characterized by subtle seizures with auditory hallucinations, infrequent nocturnal convulsions, and a good response to medications. Seizures may be triggered by voices or noises. The auditory hallucinations may be humming, clicking, ringing, or other noises. Other sensory phenomena may also occur. The interictal EEG is usually normal, though, as in the other familial disorders, IEDs may rarely be found and sporadic cases appear to have a higher incidence of epileptiform discharges (25,26).
Familial Mesial Temporal Lobe Epilepsy
The precise genetic cause of familial mesial temporal lobe epilepsy is not yet known. This disorder usually presents in adults, never before age 10 years, and therefore, it will be only briefly mentioned here. Seizures tend to be infrequent, mild, and easily controlled with medication, contrasting them to other forms of mesial temporal lobe epilepsy (27). Seizures without alteration of consciousness are more common than those with dyscognitive features. Many cases show normal imaging and EEG findings, and the presence of MR changes or interictal epileptiform activity appears to predict intractability (28).
GENETIC GENERALIZED SPIKE-WAVE EPILEPSIES
Individuals with these forms of epilepsy have normal interictal backgrounds with superimposed generalized spike-wave discharges (SWDs). These discharges will usually be 3 Hz or greater, though, at times, some slower spike-wave activity, circa 2.5 Hz may be seen, particularly in the younger child. In all cases, these occur on the backdrop of a normally developing child. There may be associated photoparoxysmal responses and some of these epilepsies will show activation of spike waves with hyperventilation. Spikes may appear more irregular and demonstrate fragmentation in the sleep record. It is not unusual to see focal features (focal slowing and focal spikes), but these will show shifting laterality from study to study.
As stated, the interictal EEG background is normal, but there may be some important exceptions: rhythmic activity may intermittently punctuate the record. This may be seen as either intermittent rhythmic theta activity, which is often in the biparietal regions (prominent in many cases of myoclonic-atonic epilepsy described by Doose) or as intermittent rhythmic delta, seen in either the occipital or the frontal regions (occipital intermittent rhythmic delta activity [OIRDA] and frontal intermittent rhythmic delta activity [FIRDA], respectively).
Generalized spike waves and other paroxysmal features found in the EEGs of individuals with these epilepsies have been known for some time to be inherited in an autosomal dominant fashion with variable penetrance so that a high proportion of family members of individuals with primary generalized epilepsy will have generalized spike waves (29,30). The clinical tendency toward epilepsy has genetic contributors as well, but these are more complex than the inheritance of the EEG trait. Although the concordance rate for primary generalized epilepsy in twin studies are high (31), the recurrence risks in first-degree relatives of patients with primary generalized epilepsies are much lower than the truly familial epilepsies with monogenic transmission (32). This argues for a more complex polygenic or oligogenic mode of inheritance.
A wide variety of epilepsies are seen and the prognosis is generally very favorable, although some will require treatment for prolonged periods. Associated epilepsies include myoclonic epilepsy in infancy (MEI), childhood absence epilepsy (CAE), epilepsy with myoclonic-atonic seizures (EMA) (as described by Doose), epilepsy with myoclonic absence, epilepsy with eyelid myoclonia (Jeavons syndrome), juvenile absence epilepsy (JAE), juvenile myoclonic epilepsy (JME), and epilepsy with generalized tonic-clonic seizures alone. Treatment is generally with broad-spectrum agents, with the exception of ethosuximide for CAE. Patients in this category will also usually not require referral to a tertiary center, unless complications arise or special circumstances present themselves.
Myoclonic Epilepsy in Infancy
The predominant seizure seen in MEI is myoclonus, as the name suggests. Infants who are developing normally have myoclonic jerks mostly of the head and proximal arms, occurring in isolation or in brief runs of recurrent jerks. The ictal EEG shows a burst of generalized or diffuse spike-wave activity with the myoclonia, otherwise the interictal EEG is normal (33) (Fig. 10.2).
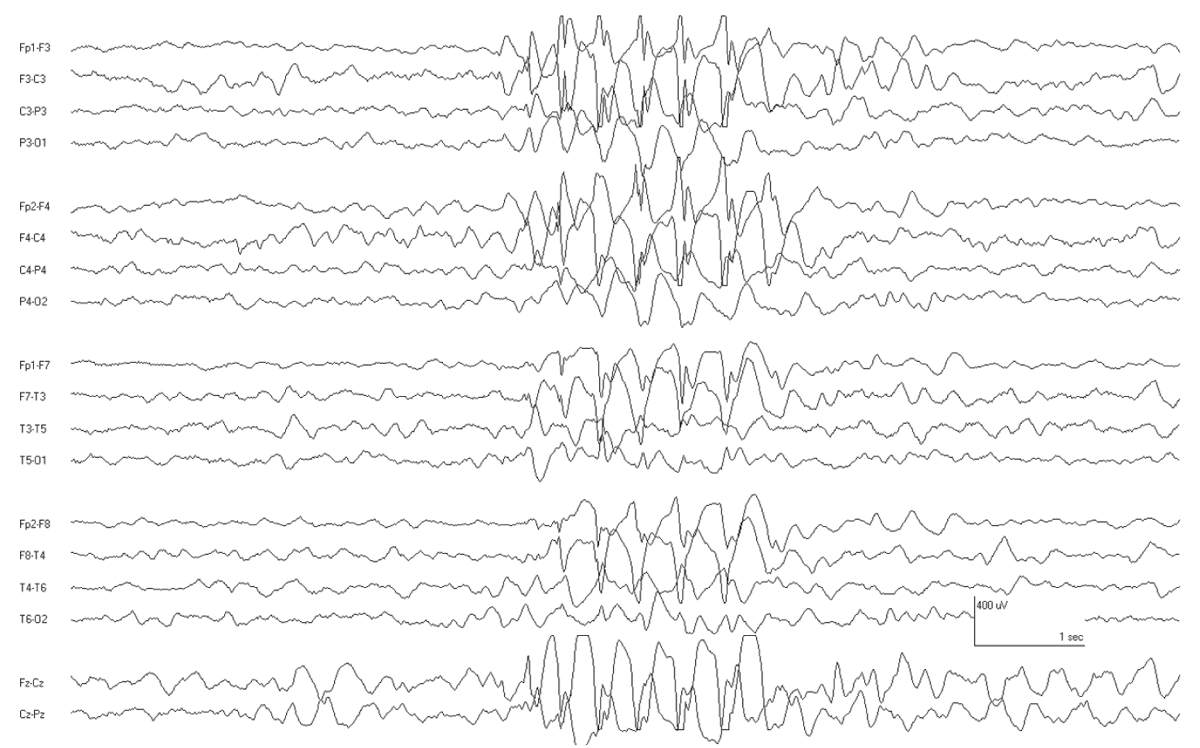
Figure 10.2: Benign myoclonic epilepsy of infancy. This infant had repeated myoclonic jerks associated with this burst of generalized spike-wave activity. Generalized spike waves are very rarely seen in infants, and this is one notable exception.
Myoclonic-Astatic Epilepsy (Doose Syndrome)
Myoclonic-astatic epilepsy may occur between late infancy and 6 years, but most often starts between 2 and 4 years. Many children will have antecedent febrile seizures. The prototypic seizure is a myoclonic-astatic attack, or now known as a myoclonic-atonic seizure (34). The seizure begins with a sudden jerk of the head or body followed by a sudden loss of tone, causing a head drop or body drop. This may easily result in injury because of the combination of the sudden propulsion from the myoclonus, the subsequent loss of postural tone, and the inability to have a protective reflex. In addition to this seizure type, atonic seizures, isolated myoclonic jerks, absence seizures, and generalized clonic-tonic-clonic seizures occur. Daytime tonic seizures are considered exclusionary by some, but nocturnal tonic seizures do occur, even in patients with excellent outcome.
The interictal EEG may show biparietal rhythmic theta activity, and this may be the only abnormality early in the course of the illness (Fig. 10.3). Bursts of 2- to 3-Hz generalized spike and polyspike-wave discharges follow and are often frequent. These occur in brief bursts, usually consisting of isolated spike waves, couplets, or triplets. The repetition rate is therefore difficult to accurately measure, but it is often variable between the bursts. Photosensitivity is common.
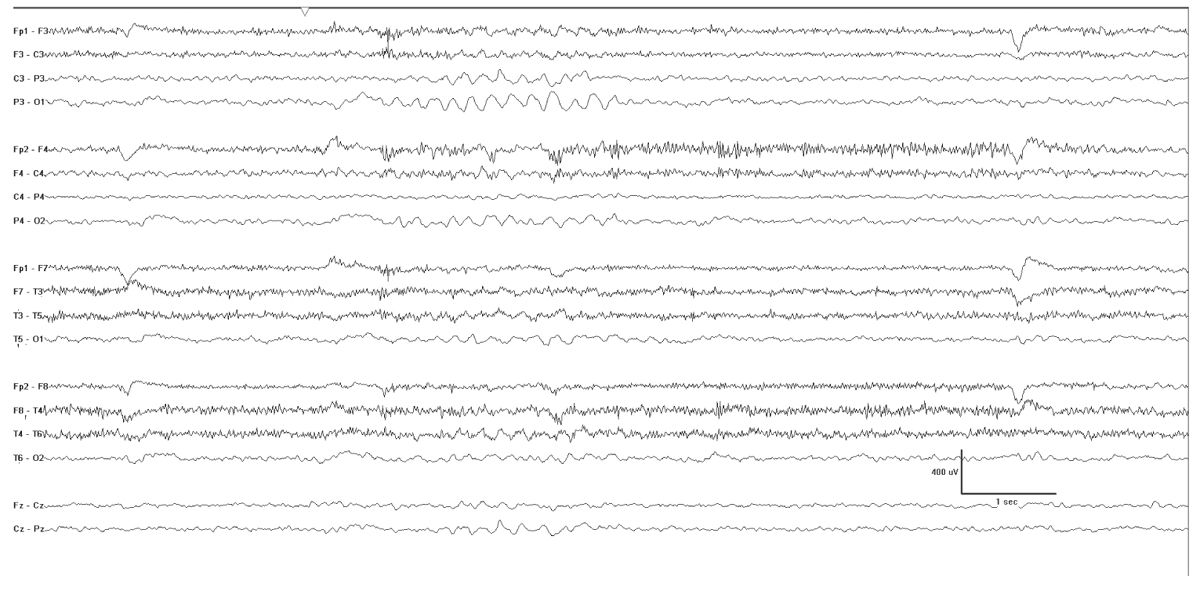
Figure 10.3: Myoclonic-atonic epilepsy as described by Doose. This tracing was obtained in a 4-year-old and was punctuated by runs of biparietal rhythmic theta activity that occurred when the patient was clearly not drowsy.
The ictal accompaniment of the myoclonic-astatic seizure is a burst of generalized polyspike-wave discharge, where the after-going slow-wave discharge appears to have some high-frequency attenuation or diminished high-frequency activity (Fig. 10.4).
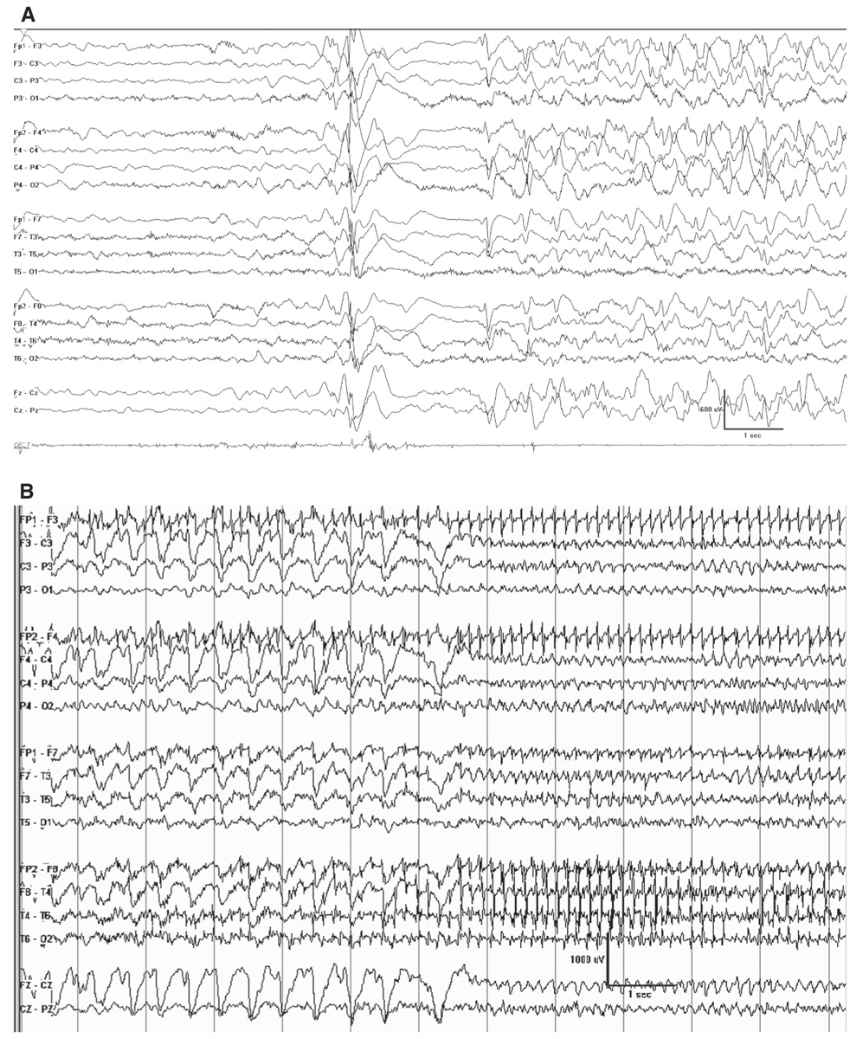
Figure 10.4: Doose syndrome. A: This youngster had a myoclonic-atonic seizure accompanied by this EEG correlate. Note the burst of generalized polyspike activity is followed by a brief epoch of relative attenuation. B: A 4-year-old with Doose syndrome had a clonic-tonic-clonic seizure, which is the typical type of convulsive event. Note the rhythmic spike waves at the start of this page corresponding to the clonic phase of the seizure, which give way to the more rapid ictal discharges corresponding with the transition to the vibratory tonic phase.
CAE generally begins between 2 and 10 years of age, with a peak in the early school-age years. From the very early observations of this form of epilepsy, it was appreciated that absences are usually frequent, with scores to hundreds of seizures in a day. This gave rise to the name pyknolepsy, signifying many seizures (35). Each individual seizure is typically brief, usually less than 10 seconds. The clinical onset and offset is abrupt, with brief impairment of consciousness associated with unresponsiveness and interruption of the ongoing activity. Some rhythmic eyeblinking may occur, but more pronounced myoclonus of the face or other body parts suggests a myoclonic form of epilepsy.
The interictal EEG is normal except that rhythmic delta activity may be seen (Fig. 10.5). The attacks are accompanied by bursts of generalized 3-Hz SWDs. These often have a frontal predominance and may have up to two spikes per complex. The frequency may slow slightly as the ictal discharge continues. Typical absence attacks often begin and end with rhythmic slowing with a more restricted topography (Fig. 10.6). Seizures are easily activated by hyperventilation, and are more frequent with lower blood glucose levels (36,37). Ethosuximide is the most effective medication with the least side effects (38).
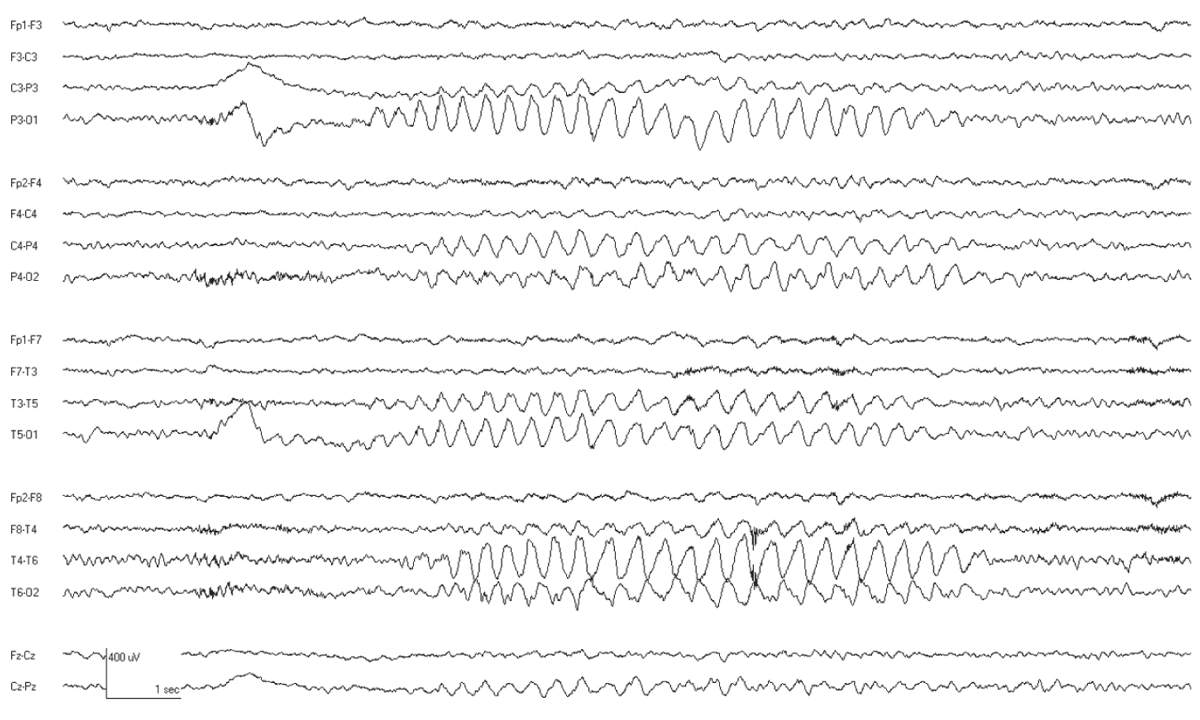
Figure 10.5: CAE. This 8-year-old child has associated OIRDA.
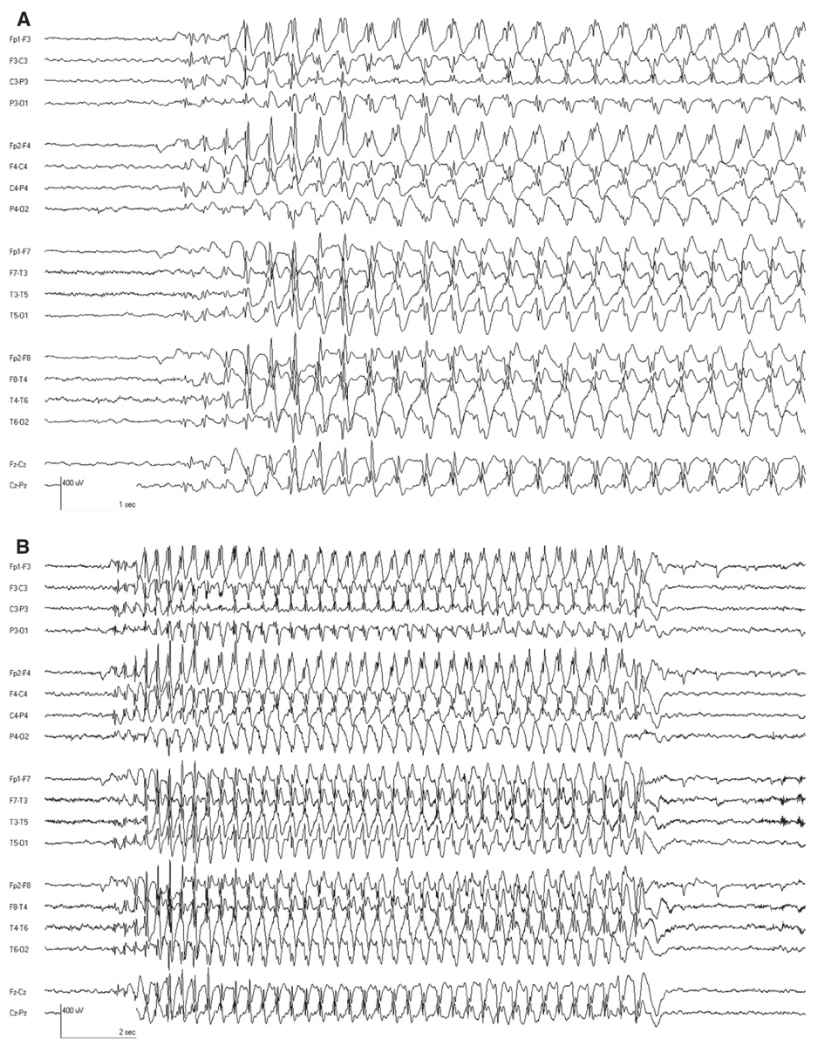
Figure 10.6: A: CAE. The classic 3-Hz SWD is evident. This was associated with unresponsiveness, confirming that it was a clinical absence attack. B: Same ictal discharge displayed with a longer page duration to reveal the whole discharge. Note how the first few discharges and last discharges are different than the main ictal pattern. (Dr. Solomon Moshé—personal comments.)
Epilepsy with Myoclonic Absences
Epilepsy with myoclonic absences is a rare form of genetic generalized epilepsy (39). It may occur at any time in infancy to adolescence, with a peak in the early school-age child, usually around 7 years (40). Myoclonic absences are characterized by tonic elevation of the arms, with repeated myoclonia of the shoulders, arms, and legs. The events may be asymmetric or even unilateral with resultant head deviation. Approximately two-thirds of children will also have tonic-clonic seizures.
The interictal EEG is normal at onset and about half of the cases show brief generalized spike-slow-wave discharges, sometimes with fragmentation. The ictal EEG shows rhythmic 3-Hz generalized polyspike-wave discharges (Fig. 10.7).
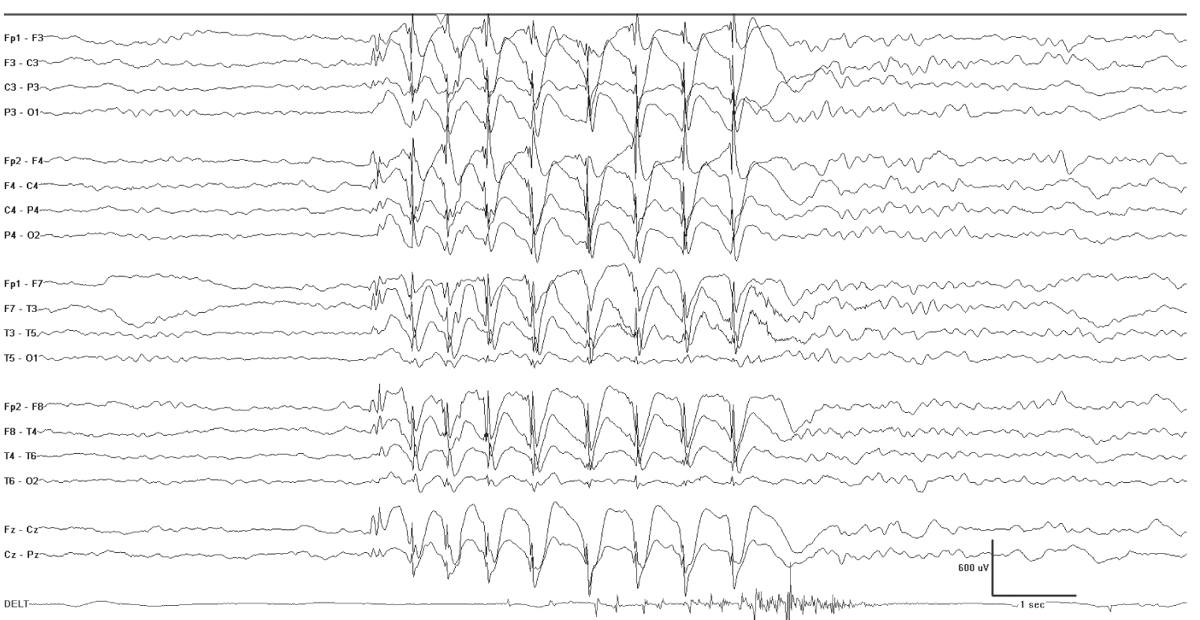
Figure 10.7: Myoclonic absence. This 6-year-old had an absence with repeated subtle head nods and arm myoclonia. Note the slight stiffening of the arm evident in the deltoid recording. There is an accompanying burst of generalized spike-wave activity.
Juvenile Absence Epilepsy
JAE and JME are both discussed in the chapter on adult epilepsy and are therefore only briefly mentioned here for completeness. This epilepsy has a peak onset between 9 and 13 years of age and most often absence attacks precede myoclonic jerks and generalized tonic-clonic seizures. The prognosis is not as favorable as CAE (41).
The interictal EEG background is normal, upon which are superimposed fragments of diffuse spikes and polyspikes. During absence attacks, there are 3- to 4-Hz generalized polyspike-wave discharges (Fig. 10.8).
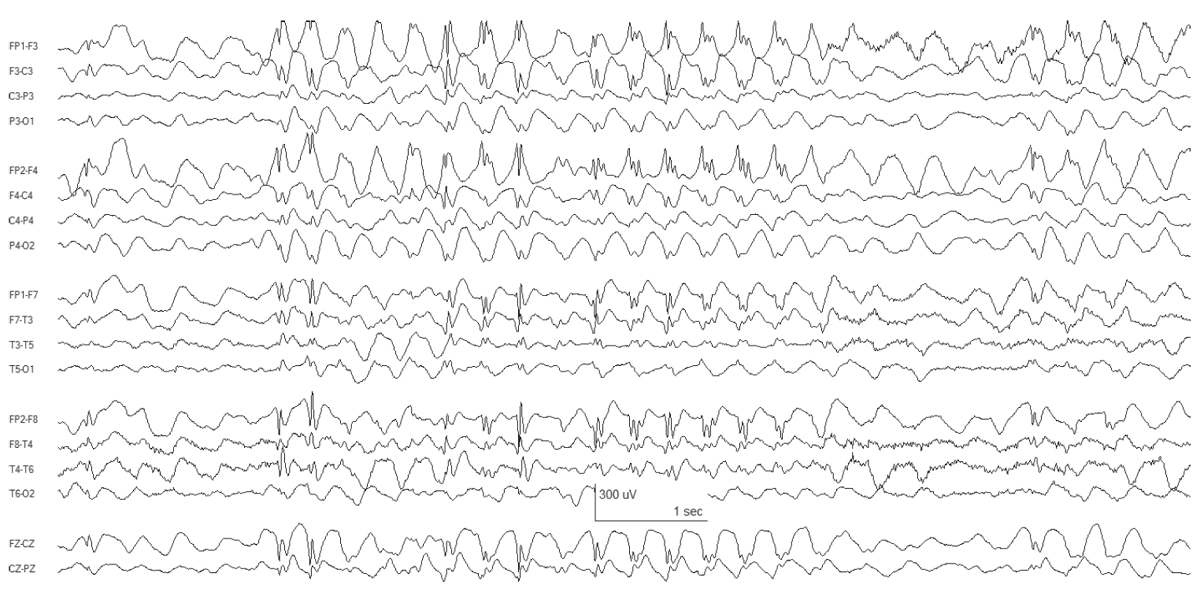
Figure 10.8: JAE. Bursts of generalized SWDs are present.
Juvenile Myoclonic Epilepsy
The history of JME has been recently reviewed (42). In 1957, Janz and Christian (43) established the essential components of JME and paid tribute to Herpin’s work dating back to 1867 with the name “Impulsiv Petit Mal.” JME is characterized by mild myoclonic, generalized clonic-tonic-clonic seizures, and absence seizures. Delgado-Escueta and Greenberg (44) searched for the genetic cause and in doing so highlighted the importance of this syndrome in the United States. Onset is typically in adolescence, with myoclonic seizures. The myoclonic seizures are usually bilateral but mild to moderate, in the sense that they may cause objects to drop but usually do not result in falls. They typically occur after awakening and may precede or lead directly into a convulsion. Seizures are aggravated by fatigue, sleep deprivation, or alcohol. Myoclonus may occur with brief absences, and myoclonic status epilepticus has been described. If untreated, generalized clonic-tonic-clonic seizures are frequent.
The interictal EEG in JME shows a normal background that is punctuated by bursts of fast generalized spike- or polyspike-wave activity with a repetition rate between 3.5- and 6-HZ spike (Fig. 10.9). During myoclonic seizures, there are 10- to 16-Hz rapid spikes with slow waves. Photosensitivity is common. Like many generalized epilepsies, fragmentation of the epileptiform discharges including clearly focal spikes can occur while awake or asleep (45).
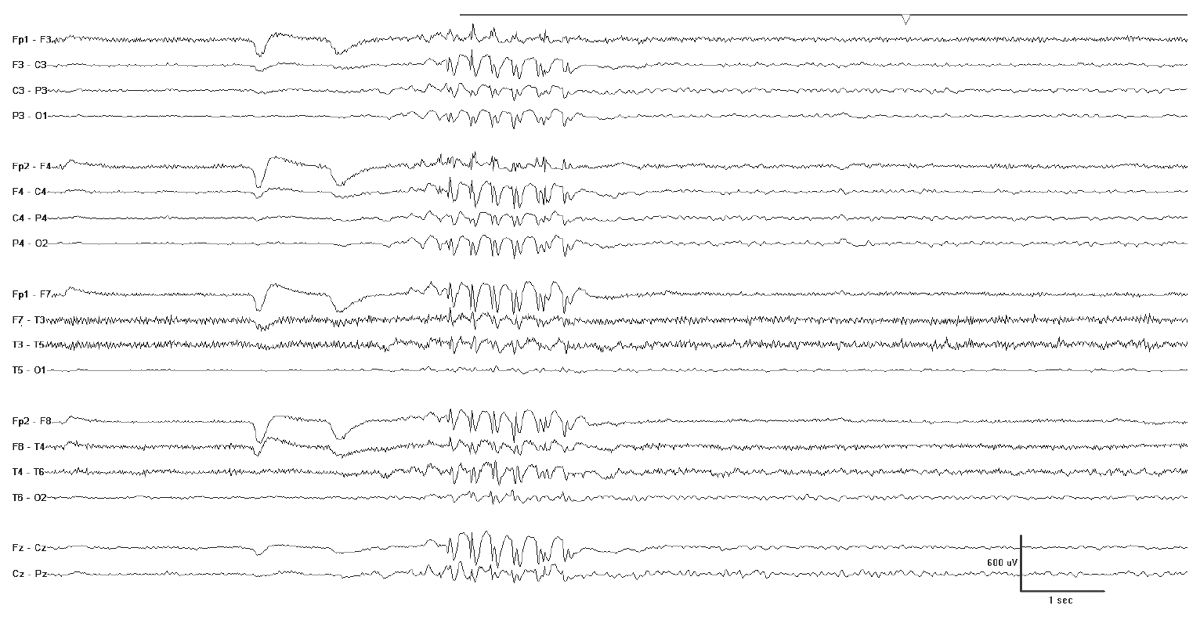
Figure 10.9: JME. Note the fast repetition rate of the generalized epileptiform discharges.
JME, similar to JAE, may require life-long treatment (46). The calcium channel, voltage-dependent beta-4 subunit (2q24), chloride channel 2 (3q26), GABA1RA (5q34), GABAR delta (1p36), and the EF-hand domain (C-terminal)-containing 1 (6p12-11) have been associated with JME.
Masquerading Conditions
There are some conditions that will produce fairly well-formed generalized spikes, but usually on an abnormal interictal EEG background. Examples of this include GLUT-1 DS (Fig. 10.10), hyperinsulinemia with hyperammonemia (Fig. 10.11), and various forms of progressive myoclonus epilepsy (Fig. 10.12). PME (e.g. EPM1, EPM2, mitochondrial cytopathies, and neuronal ceroid lipofuscionses). The background slowing and clinical histories will usually allow for easy separation of these cases, but not always. Some cases may escape detection, and therefore, the differential diagnosis should be kept broad in cases that do not show a good response to initial treatment.
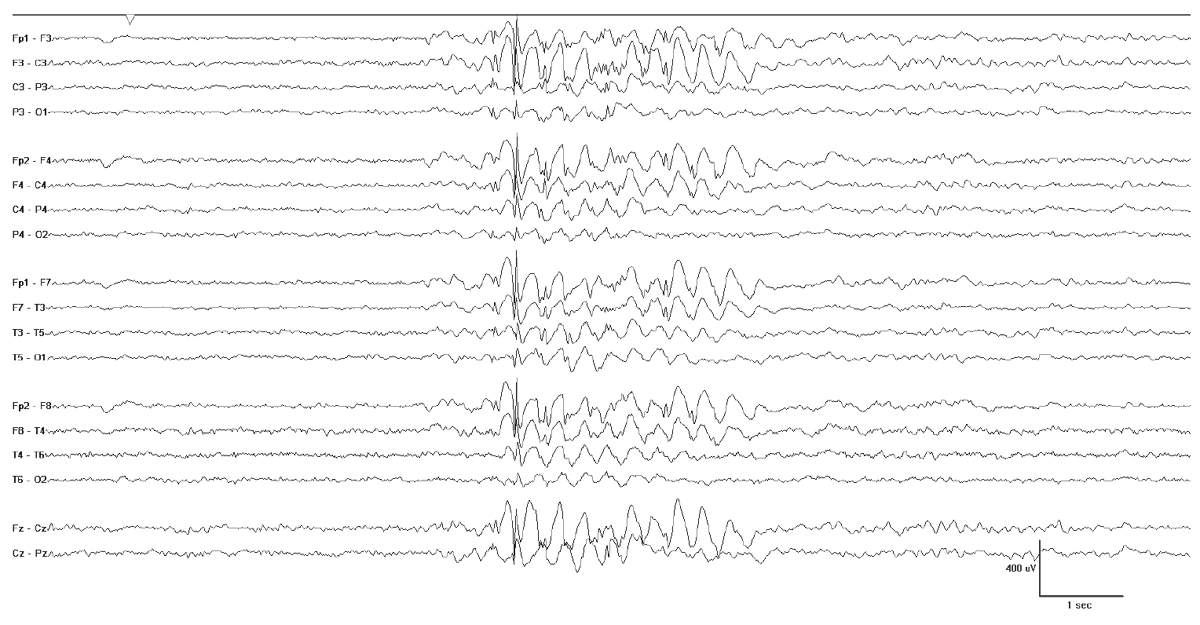
Figure 10.10: GLUT-1 DS. This 12-year-old child has bursts of generalized SWDs with a 3-Hz repetition rate. Note the slight irregular nature of the spike waves and the variability of the waveforms.
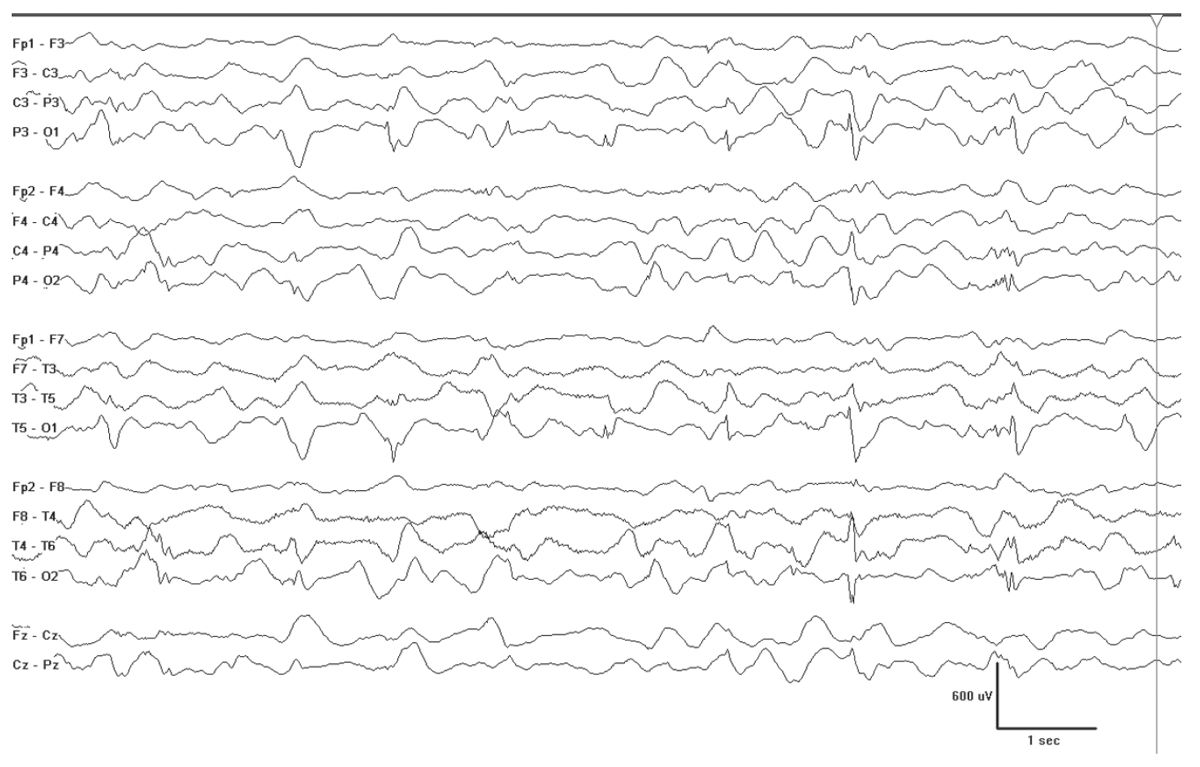
Figure 10.11: Hyperinsulinemia and hyperammonemia (HIHA). This 5-month-old has bursts of spikes that are posteriorly dominant, but ultimately will become generalized SWDs, similar to those seen in GLUT-1 DS—two conditions where the brain is not receiving adequate supply of glucose.
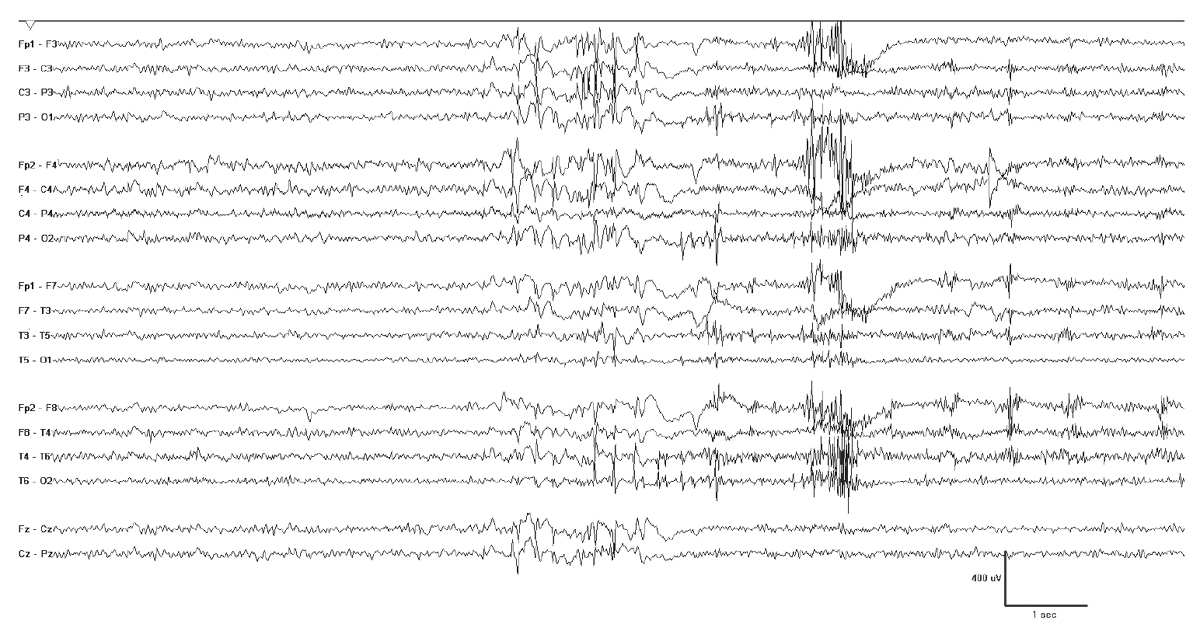
Figure 10.12: Progressive myoclonus epilepsy. This 16-year-old girl has EPM1 and her EEG shows bursts of generalized polyspike-wave discharges. These were often associated with myoclonia, but also occurred independently.
SELF-LIMITED EPILEPSIES WITH FOCAL SPIKES
The epilepsies in this second group show two common features: they have normal interictal backgrounds and they contain highly stereotyped spikes. These may be found in a single focus, in homologous regions, or in multiple foci. In some circumstances, the multifocal spikes may appear almost simultaneously. These have been referred to as “clone-like” and they appear to be driven by a primary spike generator, which is posterior located and often of the smallest amplitude (47).
It is not difficult to find the spikes and they nearly always enhance with drowsiness and sleep. Importantly, the epileptiform activity evident in sleep is identical to the activity while awake. On some occasions, the epileptiform discharges can be elicited by special maneuvers such as temporary loss of visual fixation or finger pulp tapping. There is a general pattern for the spikes to be more posterior in the younger child and to “move” more anteriorly with age. This was described by Gibbs et al. (48) and referred to as “migrating spikes.”
These epilepsies have different clinical presentations depending upon the age of presentation and the prominent location of the active focus. Panayiotopoulos uses the apt term “Benign Childhood Seizure Susceptibility Syndrome” or BCSSS for these conditions. They almost invariably have excellent outcome with regard to the cessation of seizures but may be associated with learning, behavior, or attention issues that cause havoc if not properly recognized and treated. For this reason, one might substitute “self-limited” for “benign.” Some prefer the term “epilepsy” to “seizure syndrome,” which yields a shorter term: Self-Limited Epilepsies with Focal Stereotyped Spikes (SEFS).
The genetics of the EEG trait for the prototype of this category—rolandic epilepsy (RE)—have been well worked out: spikes are inherited in an autosomal dominant fashion with variable penetrance according to age (49). A similar conclusion was reached with late-onset occipital epilepsy (50). The clinical predisposition to epilepsy, however, appears to have a relatively small genetic component as substantiated by an analysis of several twin registries (51). Based on this and other information, it has been estimated that the genetic contribution to the clinical susceptibility is small and certainly not confined to just the transmission of the EEG trait (51). This is the second EEG-epilepsy paradox: while centrotemporal spikes are seen in all individuals with RE and this trait has a genetic component, the trait alone cannot explain the genetic contribution and, moreover, the genetic contribution to the clinical susceptibility for epilepsy appears to be small (52).
Well-described self-limited epilepsies with stereotyped focal spikes include Panayiotopoulos syndrome (PS), benign epilepsy with centrotemporal spikes (BECTS) or RE, and late-onset occipital epilepsy (Gastaut type) (53). There is no described neonatal form of these epilepsies.
The spectrum of these disorders is undoubtedly larger than these well-described syndromes. We have seen children in clinical practice who have self-limited epilepsies with stereotyped spikes in other locations, including the frontal and parietal lobes, but these have not been as well characterized in the literature and apparently are not widely recognized. In addition, there can be very severe forms of almost any epilepsy, and atypical benign partial epilepsy is one such example for this group. Many highly regarded authorities also consider Landau-Kleffner syndrome and continuous spike-and-wave during sleep (CSWS) as extreme versions of these same disorders, though in 25 years of clinical practice, this author has not seen a single case transform from ordinary RE to Landau-Kleffner syndrome (LKS).
Imaging is not required for children with RE, and probably is senseless in those with multifocal stereotyped spikes, a normal background, normal neurological examination, and concordant history for a self-limited epilepsy. The challenging cases are those children with unifocal occipital or frontal stereotyped spikes. Here, it may be prudent to image to exclude the possibility of an underlying focal structural lesion, even if focal slowing and attenuation are not present.
Most children with SEFS will not require referral to a tertiary center, assuming the correct diagnosis is secured. Prophylactic treatment is generally avoided, unless seizures are particularly bothersome and recurrent, but in those children with prolonged seizures like PS, it would be wise to provide detailed instructions for an emergency plan, including administration of a rescue benzodiazepine for prolonged seizures.
Panayiotopoulos Syndrome
PS is a childhood susceptibility to focal, mainly autonomic, seizures. Children have normal physical and neuropsychological development, and peak age of onset is at 4 to 5 years of age, with a range from 1 to 14 years. The prevalence is about 13% among children, with a seizure onset between ages 3 and 6 years, and overall, about 0.2% to 0.3% of the general population of children may be affected. This figure may be much higher if children with atypical and inconspicuous presentation are included. The autonomic seizures consist of pallor, nausea, retching, and vomiting. There may be associated papillary changes and incontinence. Cardiac, breathing, and thermoregulatory alterations may occur. Cyanosis is seen, and in some severe cases, cardiac asystole has been reported (54). Most seizures are nocturnal. Many parents describe eye deviation and listlessness, but the seizures may end with hemiconvulsions or a Jacksonian march. Autonomic status epilepticus may occur, and very rarely, there may be an evolution to convulsive status epilepticus. After the postictal period resolves, the child is perfectly normal. There has been one report of an association with SCN1A mutation, but this finding was not reproduced in another family (55,56).
Neuroimaging is likewise normal. In contrast, the EEG is very useful; 90% of EEGs show multifocal stereotyped spikes and spike waves with posterior accentuation (Fig. 10.13). In follow-up EEGs, 17% had a shift of epileptiform activity from the occipital to centrotemporal areas and 3% to frontal areas (57). In PS, frontal spikes may be activated by occipital foci (58), resulting in the synchronous occipital and frontopolar spike phenomenon (59) (Fig. 10.14). The ictal EEG demonstrates rhythmic delta activity intermixed with small spikes. Onset is unilateral and most often posterior (57).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


