Chapter 36 Pathophysiologic Basis of Hibernating Myocardium
INTRODUCTION
Nuclear cardiology is increasingly used to identify viable chronically dysfunctional myocardium, and imaging is frequently the most significant factor in the decision to pursue coronary revascularization in patients with left ventricular dysfunction and ischemic cardiomyopathy. As summarized in Table 36-1, multiple pathophysiologies can account for viable dysfunctional myocardium in chronic coronary artery disease, and not all of these are the result of chronic repetitive ischemia distal to a coronary stenosis. Reversible dyssynergy has become lumped into a general category termed hibernating myocardium, but there appear to be important aspects of cellular remodeling, adaptation, and maladaptation that differ among entities responsible for viable dysfunctional myocardium. These can influence functional recovery, heart failure symptoms, and cardiovascular mortality, including sudden cardiac death. This chapter reviews our current understanding of the functional and metabolic consequences of ischemia. It also summarizes intrinsic mechanisms responsible for viable dysfunctional myocardium with an emphasis on how the physiologic changes impact the interpretation of viability imaging using single-photon emission computed tomography (SPECT), positron emission tomography (PET), and other imaging modalities.

IRREVERSIBLE ISCHEMIA AND THE EVOLUTION OF MYOCARDIAL INFARCTION
Evolution of Acute Myocardial Injury
The temporal evolution and extent of irreversible myocardial injury after coronary occlusion is quite variable and strongly dependent on the magnitude of residual coronary flow, the hemodynamic determinants of oxygen consumption, and any endogenous protection afforded by preconditioning.1,2 Reimer and Jennings demonstrated that in the absence of significant collaterals, irreversible injury begins approximately 20 minutes after coronary occlusion3 and progresses in a wavefront from the subendocardium to the subepicardium (Fig. 36-1).4 This susceptibility of the subendocardium to injury reflects its higher oxygen consumption and the redistribution of collateral flow to the outer layers of the heart by the compressive determinants of flow at reduced coronary pressure.5 In experimental infarction, the entire subendocardium is irreversibly injured within 1 hour of occlusion, and the transmural progression of infarction is largely complete within 4 to 6 hours. Factors that increase myocardial oxygen consumption (e.g., tachycardia, hypertension) or reduce oxygen delivery (e.g., anemia, hypotension) accelerate the progression to irreversible injury. In contrast, repetitive reversible ischemia prior to occlusion can limit irreversible injury through a preconditioning effect.1
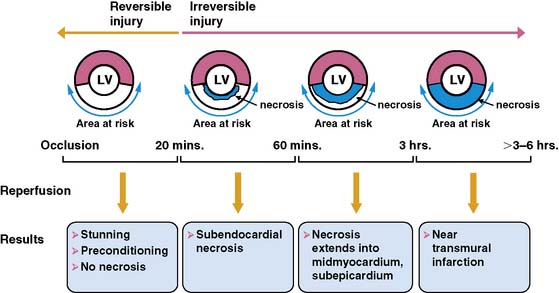
Residual Coronary Flow Limits Infarction
The magnitude of residual coronary flow, either due to subtotal coronary occlusion or the presence of intercoronary collaterals, is the most important determinant of the actual time course of irreversible injury. Ultimate infarct size as a percent of the area at risk of ischemia during a total occlusion is inversely related to collateral flow.6 Subendocardial flow of as little as 30% of resting values can prevent infarction during ischemic periods exceeding an hour. More moderate subendocardial ischemia (e.g., flow reduced by 50%) can persist for hours without resulting in significant irreversible injury.7 This phenomenon explains how the signs and symptoms of ischemia can be present for prolonged periods without producing significant myocardial necrosis. It also accounts for the observation that late coronary reperfusion can salvage myocardium beyond the 6-hour limit predicted from experimental models.
FUNCTIONAL CONSEQUENCES OF ACUTE REVERSIBLE ISCHEMIA
Fortunately, reversible ischemia is considerably more common than irreversible injury. The functional consequences of acute ischemia are summarized in Figure 36-2. Transient coronary occlusion as a result of coronary vasospasm or temporary thrombosis produces supply-mediated ischemia similar to that present at the onset of infarction (vide supra). Upon reperfusion, regional function can remain depressed as a result of myocardial stunning (vide infra). Demand-induced ischemia develops when regional perfusion is unable to sufficiently increase in response to increases in myocardial oxygen demand; this predominantly affects the subendocardium. Interestingly, these two mechanisms of acute ischemia have fundamentally different effects on myocardial diastolic relaxation, with supply-mediated ischemia increasing LV compliance and demand-induced ischemia reducing it. Because of the temporal delay in the development of angina, many episodes of acute myocardial ischemia associated with ST-segment depression are symptomatically silent. Very brief episodes of ischemia (as reflected by more sensitive indices such as regional dysfunction or elevations in end-diastolic pressure) can even be electrocardiographically silent.
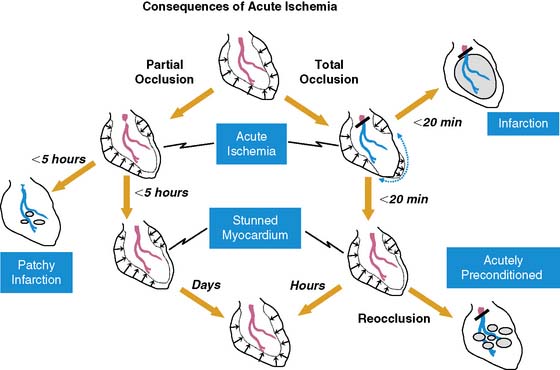
Stunned Myocardium
There is a temporal delay in the recovery of function after single episodes of ischemia lasting more than 2 minutes. This regional dysfunction occurs despite the restoration of perfusion, with the time course of recovery determined by the duration and severity of the antecedent ischemia. Heyndrickx et al. were the first to demonstrate that regional dysfunction could persist for hours following a 15-minute occlusion, despite complete reperfusion (Fig. 36-3),8 a phenomenon that Kloner and Braunwald subsequently called stunned myocardium.9 The physiologic hallmark of stunned myocardium is a dissociation of the usual close relation between subendocardial flow and function, where regional dysfunction is present despite normal levels of resting perfusion. Stunned myocardium can also occur after demand-induced ischemia. For example, in the presence of a coronary stenosis, exercise can result in depressed regional function that persists for hours after perfusion is restored, and repetitive ischemia can lead to cumulative stunning.10 From a clinical perspective, the identification of stunned myocardium is important, since unlike other dysfunctional states, regional function will spontaneously normalize in the absence of recurrent ischemia. Furthermore, contractile reserve is invariably present with inotropic stimulation (e.g., dobutamine infusion), usually normalizing regional function (see Table 36-1). In contrast to animal models, acutely stunned myocardium in patients is not always a “pure entity” and frequently coexists with irreversibly injured and viable, chronically dysfunctional myocardium (i.e., chronically stunned and hibernating myocardium, vide infra).
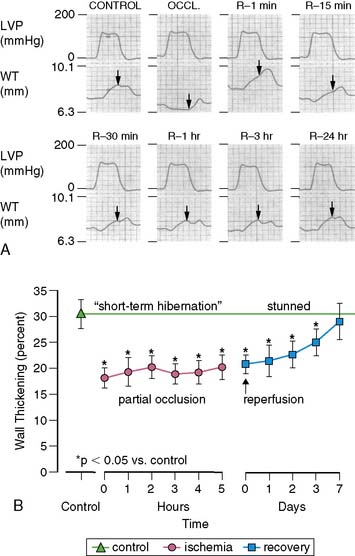
(A, Modified from Heyndrickx et al.84 and republished with permission from the American Physiological Society. B, From Matsuzaki M, Gallagher KP, Kemper WS, et al: Sustained regional dysfunction produced by prolonged coronary stenosis: Gradual recovery after reperfusion, Circulation 68:170–182, 1983. Reprinted with permission.)
Short-Term Hibernation During Prolonged Moderate Ischemia
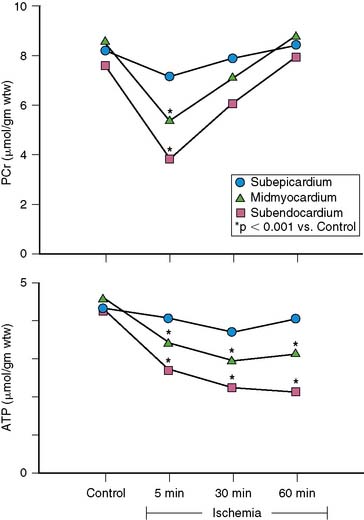
When coronary pressure distal to a stenosis falls below the lower limit of autoregulation, flow reserve is exhausted, resulting in subendocardial ischemia during which reductions in subendocardial flow are closely coupled to reductions in regional contractile function.11 When moderate ischemia persists, the close matching between perfusion and contraction results in a new steady-state with reduced regional oxygen consumption and energy utilization, a phenomenon termed short-term hibernation.7,11 Despite persistent hypoperfusion, the balance between oxygen supply and demand is reestablished, as reflected by regeneration of creatine phosphate and ATP as well as the resolution of lactate production (Fig. 36-4).12 Importantly, this ability to restore an energetic balance allows myocyte viability to be maintained for a much longer period of time than when flow is absent. Reperfusion of short-term hibernation also leads to stunning, which may take up to a week to resolve (see Fig. 36-3).13
While short-term hibernation was originally hypothesized to be the mechanism of chronic hibernating myocardium, it is now clear that short-term hibernation is an extremely tenuous condition, with small increases in myocardial oxygen demand precipitating recurrent ischemia and a rapid deterioration in function and metabolism.6 Thus, the reversible nature of short-term hibernation is limited by the severity and duration of ischemia, with irreversible injury frequently occurring after more than 12 to 24 hours.14
PATHOPHYSIOLOGY OF CHRONIC REPETITIVE ISCHEMIA
Over 60% of patients with ischemic cardiomyopathy have regional dysfunction as a result of repetitive episodes of myocardial ischemia. This dysfunction is part of a pathophysiologic continuum from chronically stunned myocardium with normal resting perfusion to chronic hibernating myocardium with reduced resting perfusion, which appears to be related to the functional significance of the underlying coronary disease and therefore its propensity to cause ischemia (Fig. 36-5).15,16 The distinguishing physiologic features differentiating chronically stunned from hibernating myocardium are described in the following sections.
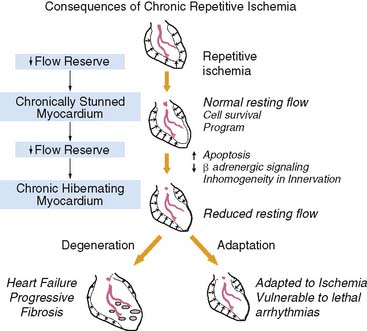
Figure 36-5 Functional consequences of repetitive ischemia. With the progression in severity of a stenosis, coronary flow reserve decreases, and ischemia becomes more frequent. Repetitive ischemia initially leads to chronic preconditioning against infarction and stunning (see Fig. 36-2), which maintains resting function and prevents the development of infarction. As stenosis severity and the frequency of spontaneous ischemia increase further, there is a gradual progression from contractile dysfunction with normal resting flow (chronically stunned myocardium) to contractile dysfunction with depressed resting flow (hibernating myocardium). This transition is related to the physiologic significance of a coronary stenosis and can occur in a time frame as short as 1 week. Alternatively, it can develop chronically and progress in the absence of symptomatic ischemia. The cellular responses during the progression to chronic hibernating myocardium are quite variable. While some patients exhibit little cell death and fibrosis (Adaptation), others develop a picture characterized by degenerative changes that include progressive fibrosis and myocyte death that is difficult to distinguish from subendocardial infarction (Degeneration). The factors that determine the variable progression have not been resolved.
(Modified from Canty JM Jr: Coronary blood flow and myocardial ischemia. In Libby P, Bonow RO, Mann DL, et al (eds): Braunwald’s Heart Disease, 8th ed. Philadelphia: Elsevier, 2007, pp 1167–1194.)
Chronically Stunned Myocardium
Basic studies have clearly shown that repetitive episodes of myocardial ischemia with17–19 and without a residual stenosis20,21 can result in chronic regional dysfunction with normal resting perfusion. In experimental animal models with a progressive stenosis, chronically stunned myocardium precedes the development of hibernating myocardium.18,22 Although resting flow is normal in chronically stunned myocardium, the regional uptake of the glucose analog, fluorine-18 2-fluorodeoxyglucose (FDG), is variably increased and appears to be related to the physiologic severity of a coronary stenosis. For example, pigs chronically instrumented with a fixed-diameter left anterior descending (LAD) coronary artery stenosis have regional dysfunction with normal resting perfusion at both 1 and 2 months after initial instrumentation (Fig. 36-6).18 Regional FDG uptake is initially normal, but it becomes increased as the reduction in flow reserve progresses, and this persists with the transition to hibernating myocardium.
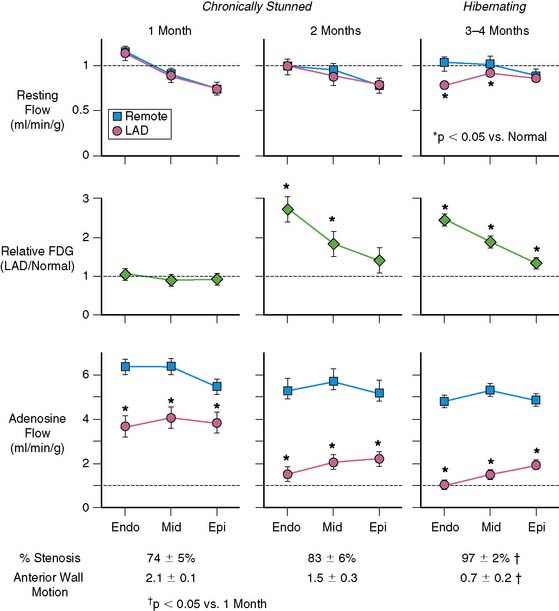
From a clinical perspective, it is important to recognize that viable, chronically dysfunctional myocardium with normal resting perfusion is much more common than hibernating myocardium,23,24 and it can develop independently of ischemia. For example, in patients with ischemic cardiomyopathy, regional dysfunction with normal resting perfusion may occur distant from a large dysfunctional region (viable or infarcted) as a result of pathologic remodeling. Dysfunction in this remodeled myocardium will not improve with regional revascularization, since it is not a direct result of ischemia. Thus, normal resting perfusion does not necessarily translate into reversible dyssynergy, and it is not surprising that noninfarcted but remodeled segments sometimes have persistent dysfunction after coronary revascularization.25
Chronic Hibernating Myocardium
Hibernating myocardium is characterized by contractile dysfunction with reduced resting flow, in the absence of acute ischemia or significant necrosis. It is common in patients with ischemic cardiomyopathy,26 where it frequently coexists with nontransmural infarction and can also occur in the absence of heart failure or global LV dysfunction. The best clinical example of isolated hibernating myocardium is in patients with chronic coronary occlusions and collateral-dependent myocardium (Fig. 36-7).27,28 A similar constellation of findings has been reproduced in pigs in the absence of infarction or heart failure, which has facilitated the investigation of the underlying adaptations to ischemia that result in hibernating myocardium.22 Studies examining these key physiologic adaptations are summarized later.
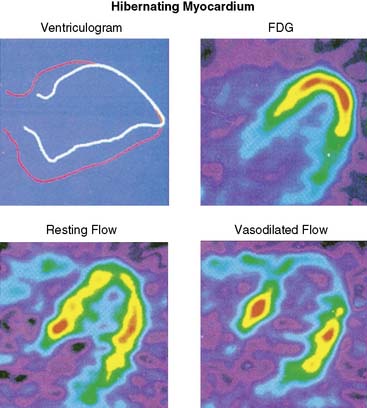
The unpredictable progression of clinical coronary disease precludes defining the temporal evolution of hibernating myocardium in patients, and it was originally thought that hibernating myocardium arose from a primary reduction in flow in a fashion similar to experimental models of prolonged moderate ischemia and short-term hibernation.7 While this is a plausible mechanism for the development of hibernating myocardium in association with an acute coronary syndrome, more recent studies have highlighted the role of the physiologic severity of a coronary stenosis and repetitive ischemia as the major determinants of the progression from stunned to hibernating myocardium. This paradigm developed from animal studies with a slowly progressive stenosis that clearly demonstrated that regional dysfunction with normal resting flow (chronically stunned myocardium) preceded the development of hibernating myocardium, with the down-regulation in resting flow associated with a critical impairment in subendocardial flow reserve (see Fig. 36-6).15,18,22,29 This progression is not merely a function of time, since the transition from chronically stunned to hibernating myocardium can be experimentally produced in as little as 1 week after placement of a critical stenosis which exhausts coronary flow reserve.19 A common conclusion of all serial studies is that reduced resting flow in hibernating myocardium is a result rather than a cause of the contractile dysfunction. As will be discussed, the down-regulation of resting flow in hibernating myocardium appears to be a physiologic marker reflecting numerous intrinsic metabolic adaptations of the heart to repetitive episodes of ischemia.
Metabolic and Energetic Adaptations to Chronic Ischemia
Myocardial Glucose Uptake
Acute myocardial ischemia is obviously associated with acute reductions in oxidative metabolism and an increased dependence on anaerobic glycolysis. This is facilitated by the mobilization of glycogen stores, as well as an increase in myocardial glucose uptake secondary to the translocation of the glucose transporter GLUT4 from intracellular vesicles. This underlies the basis of “hot spot” imaging with the glucose analog FDG for exercise-induced ischemia and acute coronary syndromes30 and is likely responsible for the enhanced FDG uptake in viable dysfunctional myocardium following dobutamine infusion.31
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


