Pathophysiology of Myocardial Ischemia
The major cause of acute myocardial infarction (MI) is coronary atherosclerosis with superimposed luminal thrombus, which accounts for more than 80% of all infarcts. MIs resulting from nonatherosclerotic diseases of the coronary arteries are rare. In past decades, there have been several trends regarding the epidemiology and outcome of patients hospitalized with acute MI. Over the time span from 1975 to 2001, patients became significantly older, were more likely to be women, and were more likely to receive effective cardiac medications. Despite a greater prevalence of comorbidities, hospital survival rates have improved over time.1
The normal function of the heart muscle is supported by high rates of myocardial blood flow, oxygen consumption, and combustion of fat and carbohydrates (glucose and lactate). Under normal aerobic conditions, cardiac energy is derived from fatty acids, supplying 60% to 90% of the energy for adenosine triphosphate (ATP) synthesis (Fig. 57–1). The rest of the energy (10%-40%) comes from oxidation of pyruvate formed from glycolysis and lactate oxidation. Almost all of the ATP formed comes from oxidative phosphorylation in the mitochondria; only a small amount of ATP (<2%) is synthesized by glycolysis. Approximately two-thirds of the ATP used by the heart goes to contractile shortening, and the remaining third is used by sarcoplasmic reticulum Ca2+ ATPase and other ion pumps.
Figure 57–1.
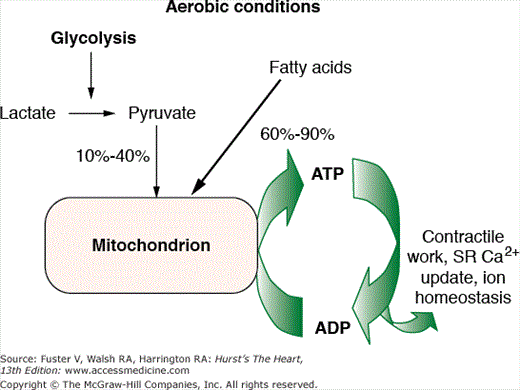
Cardiac energy metabolism under normal aerobic conditions. Fatty acids are the primary source of energy for the heart, supplying 60% to 90% of the energy for adenosine triphosphate (ATP) synthesis. The balance (10%-40%) comes from the oxidation of pyruvate formed from glycolysis and lactate oxidation. Almost all of the ATP formation comes from oxidative phosphorylation in the mitochondria; only a trivial amount of ATP (<2% of the total) is synthesized by glycolysis. ADP, adenosine diphosphate; SR, sarcoplasmic reticulum. Reproduced with permission from Stanley WC. Changes in cardiac metabolism: a critical step from stable angina to ischemic cardiomyopathy. Eur Heart J Suppl. 2001;3(suppl O):O3.
Sudden occlusion of a major branch of a coronary artery shifts aerobic or mitochondrial metabolism to anaerobic glycolysis within seconds of reduced arterial flow. Myocardial ischemia primarily affects mitochondrial metabolism, resulting in a decrease in ATP formation by shutting off oxidative phosphorylation. The reduced aerobic ATP formation stimulates glycolysis and an increase in myocardial glucose uptake and glycogen breakdown (Fig. 57–2). Decreased ATP inhibits Na+, K+-ATPase, increasing intracellular Na+ and Cl, leading to cell swelling. Derangements in transport systems in the sarcolemma and sarcoplasmic reticulum increase cytosolic Ca2+, inducing activation of proteases and alterations in contractile proteins. Pyruvate is not readily oxidized in the mitochondria, leading to the production of lactate, a decrease in intracellular pH, and a reduction in contractile function. The decrease in pH also leads to greater ATP requirement to maintain Ca2+ homeostasis.2
Figure 57–2.
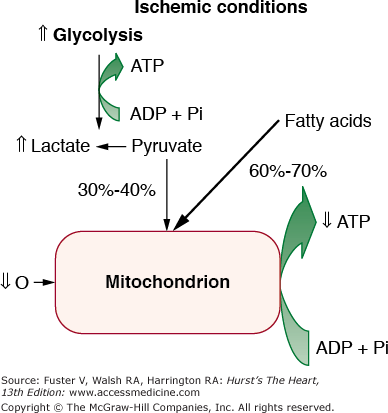
Cardiac energy metabolism during ischemia of moderate severity (~40% of normal blood flow). The up and down arrows indicate the changes compared with normal conditions. Relative to aerobic conditions, ischemia results in an increase in glycolysis without an increase in the rate of pyruvate oxidation, thus causing lactate to accumulate in the cell. Despite accelerated glycolysis and lactate production, the relatively high rate of residual oxygen consumption is fueled primarily by the oxidation of fatty acids. ADP, adenosine diphosphate; ATP, adenosine triphosphate; Pi, inorganic phosphate. Reproduced with permission from Stanley WC. Changes in cardiac metabolism: a critical step from stable angina to ischemic cardiomyopathy. Eur Heart J Suppl. 2001;3(suppl O):O5; Fig. 4.
Electron microscopic alterations result from reversible and irreversible myocardial ischemic injury and are similar across animal species, with some variation in time course. Reversibly injured myocytes are edematous and swollen from the osmotic overload. The cell size is increased with a decrease in the glycogen content.3-5 The myocyte fibrils are relaxed and thinned; I-bands are prominent secondary to noncontracting ischemic myocytes.6 The nuclei show mild condensation of chromatin at the nucleoplasm. The cell membrane (sarcolemma) is intact, and no breaks can be identified. The mitochondria are swollen, with loss of normal dense mitochondrial granules and incomplete clearing of the mitochondrial matrix but without amorphous or granular flocculent densities (Fig. 57–3). Irreversibly injured myocytes contain shrunken nuclei with marked chromatin margination. The two hallmarks of irreversible injury are cell membrane breaks and mitochondrial presence of small osmiophilic amorphous densities.7 The densities are composed of lipid, denatured proteins, and calcium.8 The cell membrane breaks are small and are associated with subsarcolemmal blebs of edema fluid. (see Fig. 57–3B).
Figure 57–3.

A. Sequential changes within mitochondria with varying time intervals of myocardial ischemia. At 20 minutes of ischemia, there is mild mitochondrial swelling. Matrix space between cristae show disorganization. At 40 minutes of ischemia, there is greater mitochondrial swelling, and prominent amorphous matrix densities are present, which indicate irreversible injury. With a longer duration of coronary occlusion, mitochondria show larger amorphous matrix densities, and they also become more numerous. On reperfusion, both amorphous and granular densities are seen. Granular densities, however, seem larger and more fully developed. Adapted with permission of American Heart Association from Jennings and Ganote.7 B. Electron micrographs showing progressive changes in mitochondria as a result of ischemia in the canine model. Panel a. Mitochondria showing reversible changes of ischemia after 10 minutes of coronary occlusion and reperfusion: mitochondria are swollen, there is clearing of mitochondrial matrix, and some cristae show disorganization. Panel b. Similar changes as in panel a with only one of the mitochondria showing amorphous matrix densities (arrowhead) after 90 minutes of ischemia. Panel c. Note the presence of large amorphous matrix densities (arrowheads) in two of the three mitochondria in a dog with 120 minutes of coronary occlusion. Panel d. Ischemic myocyte with mitochondria containing multiple large amorphous matrix densities (arrowheads) after 3 days of permanent coronary occlusion. Note the break in the plasma lemma (arrow). Reproduced with permission from the American Heart Association from Virmani, Forman, Kolodgie.5
Irreversible ischemic injury is characterized by a variety of processes involving the sarcolemmal membrane eventuating in its disruption and cell death. Increased cytosolic Ca2+ and mitochondrial impairment cause phospholipase activation and release of lysophospholipids and free fatty acids, which are incorporated within the cell and damaged by peroxidative damage from free radicals and toxic oxygen species. Cleavage of anchoring cytoskeletal proteins and progressive increases in cell membrane permeability result in physical disruption and cell death.8
There are two basic types of cell death: oncosis, which results in cell swelling and is associated with ischemic damage, and apoptosis, which results from cell shrinkage. In general, whereas oncosis results from events exogenous to the cell, apoptosis is programmed and is ATP dependent. Oncosis is independent of energy supply and caspase activity. Because apoptosis is energy dependent, it has not been classically implicated in ischemic myocyte death; however, experiments have suggested that apoptosis is involved in the first hours of ischemic injury, especially during reperfusion. The detection of apoptosis depends on identifying the final outcome of the apoptotic pathway (double-stranded DNA fragmentation), but there are issues of specificity associated with detection of these fragments by the most commonly used technique, terminal deoxynucleotidyl transferase–mediated biotinylated deoxyuridine triphosphate nick-end labeling (TUNEL). Other features of apoptosis that can be assayed include activation of cytosolic aspartate-specific cysteine proteases, or caspases; cytochrome C release in mitochondria; and selective alteration of cell membranes with an increased expression of phosphatidylserine in the outer membrane, with preservation of selective membrane permeability (generally accomplished by annexin-V labeling). Various of these alterations have been described in ischemic myocardium, but the current consensus is that apoptosis and oncosis proceed together in ischemic myocytes, with oncosis dominating, especially in end stages.8
Autophagic, or ubiquitin-related, cell death is characterized ultrastructurally by autophagic vacuoles, cellular degeneration, and nuclear disassembly.9 Similar to apoptosis, autophagic cell death is energy dependent, but in contrast to apoptosis, is caspase independent. The formation of autophagic vacuoles involves posttranslational modification of proteins by linkage to numerous ubiquitin molecules, making them susceptible to proteasomal digestion. A variety of proteins, including cathepsin D, cathepsin B, heat shock cognate protein Hsc73, beclin 1, and the processed form of microtubule-associated protein 1 light chain 3, are known to mediate autophagy. Autophagic cell death has been described in hypertrophied and failing myocardium and has been identified as increasing in hibernating myocardium.10 The initiation of autophagy does not always result in death of the cell because autophagy may be responsible for the turnover of unnecessary or dysfunctional organelles and cytoplasmic proteins. In a model of repetitive ischemia in pigs, autophagic cell death has been demonstrated to occur later than apoptosis and, inversely, suggested a protective effect against ischemia-induced apoptosis.11
Myocardial ischemia occurs when oxygen supply does not meet myocardial demand, and necrosis or infarction occurs when ischemia is severe and prolonged. Although biochemical and functional abnormalities begin almost immediately at onset of ischemia, severe loss of myocardial contractility occurs within 60 seconds, other changes take a more protracted course; for example, the loss of viability (irreversible injury) takes at least 20 to 40 minutes after total occlusion of blood flow.
Two zones of myocardial damage occur: a central zone with no flow or very low flow, and a zone of collateral vessels in a surrounding marginal zone. The survival of the marginal zone depends on the level of ischemia and the duration of ischemia. In autopsy hearts, the size of the ischemic zone surrounding an acute MI is associated with increased apoptosis and degree of occlusion of the infarct-related artery.12 The extent of coronary collateral flow is one of the principal determinants of infarct size. Indeed, at autopsy, it is common to see chronic total coronary occlusion and an absence of MI in the distribution of that artery. Absence of myocardial ischemia (revealed by electrocardiographic changes or angina during transient coronary balloon occlusion) is associated with presence of well-developed collateral vessels, suggesting that the patients with well-developed collateral vessels have a low risk of developing acute MI upon abrupt closure of the culprit coronary artery.13 Collaterals are better developed in patients with angina and in younger individuals than in older patients with acute infarcts.14 Because infarct size is an important determinant of survival as well as development of congestive heart failure, efforts have been directed to limit infarct size by early reperfusion, reduction of myocardial oxygen demand, and prevention of reperfusion injury. In 1971, Page et al15 showed that infarcts 40% of less of the left ventricle are predictors of cardiogenic shock and death.
Reimer and Jennings, initially in 1979, showed that if a canine coronary artery was occluded for 15 minutes, for 40 minutes, for 3 hours, or permanently for 4 days, myocardial necrosis progressed as a “wave front phenomenon” (Fig. 57–4A).16,17 The extent of myocardial necrosis therefore depended on the duration of coronary occlusion. After only 15 minutes of occlusion, no infarct occurred. At 40 minutes, the infarct was subendocardial, involving only the papillary muscle, resulting in 28% of the myocardium at risk. At 3 hours after coronary artery occlusion and reperfusion the infarct was significantly smaller compared with nonreperfused permanently occluded infarct (62% of area at risk). The infarct size was the greatest in permanent occlusion, becoming transmural involving 75% of the area at risk (Fig. 57–4B).6 In the dog model, it is impossible to achieve 100% infarction of the area at risk because of species-related native collaterals. In humans, it has been shown that approximately 40% of patients with acute MI have well-developed collateral circulation.14
Figure 57–4.
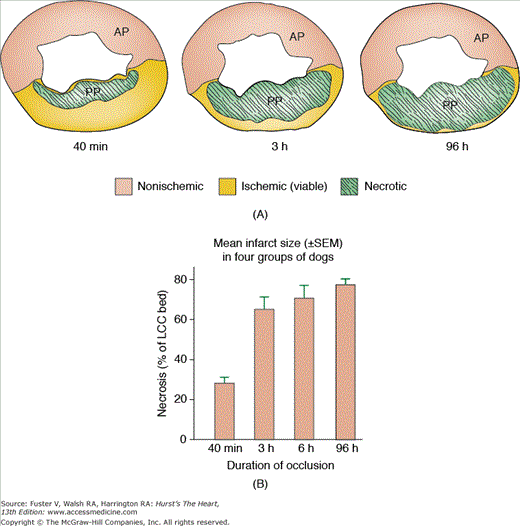
A. Progression of cell death versus time after experimental occlusion of the left circumflex coronary artery in the dog. Necrosis occurs first in the subendocardial region of the myocardium. With extension of the occlusion time, a wavefront of cell death moves from the subendocardial zone across the wall to involve progressively more of the transmural thickness of the ischemic zone. B. Infarct size variation with increasing duration of coronary occlusion. The infarct size dramatically increases from 40 minutes to 3 hours; however, there is very little increase between 3 and 6 hours and between 6 and 96 hours of coronary artery occlusion. AP, anterior; LCC, left circumflex coronary bed; PP, posterior. Reproduced by permission from Reimer and Jennings.16
Other than the presence of collateral circulation, factors that influence infarct size include preconditioning, which may greatly reduce infarct size, and reperfusion. However, there is a balance between benefits of reperfusion in reducing infarct size and reperfusion injury, which is dependent on the time of onset. In general, if ischemic myocardium is reperfused early, the degree of myocardial salvage greatly exceeds damage from free radicals and calcium loading caused by reperfusion. These positive functional consequences of reperfusion are most beneficial within the initial 12 hours after occlusion in humans.
It was documented in the late 1970s that transmural infarcts may increase in size for weeks after the initial event, and the degree of this expansion is associated with a decrease in survival rate.18 The processes involved in postinfarction ventricular dilatation are known as ventricular remodeling. In general, transmural extent of necrosis is a major determinant of infarct expansion (remodeling) based on large infarct size and the persistence of the occlusion. The preservation of islands of viable myocardium in the subepicardial regions is associated with decreased remodeling or infarct expansion. Other factors that are implicated in reduced ventricular remodeling include microvascular integrity19 and initial ventricular compliance, as measured by mitral deceleration time.20 The effect of reperfusion on ventricular remodeling is clear as far as early reperfusion is concerned because there are definite benefits in reducing infarct size and expansion. The benefits of late reperfusion beyond myocardial salvage are unclear. It has been demonstrated that remodeling is affected by the presence of viable zones after successful late percutaneous coronary intervention (PCI).21 In general, the mechanisms of ventricular remodeling are poorly understood because different techniques have been used to assess myocardial viability in human subjects, animal studies, and postmortem specimens. The release of matrix metalloproteinases is now being linked to remodeling.
The no-reflow phenomenon was originally described by Kloner et al22 in 1974 in an experimental canine model of MI. They demonstrated homogenous distribution of thioflavin S dye after 40 minutes of ischemia and reperfusion. However, after 90 minutes of ischemia, areas of no-reflow were identified mainly in the subendocardial regions as zones not staining with thioflavin S. By electron microscopy, they showed swollen endothelial protrusions and membrane-bound intraluminal bodies, which obstructed the capillary lumen and resulted in plugging of the capillaries by red blood cells, neutrophils, platelets, and fibrin thrombi. The areas not stained by thioflavin S were characterized by low regional myocardial blood flow.
Angiographic no-reflow is also a strong predictor of major cardiac events, similar to congestive heart failure, malignant arrhythmias, and cardiac death, after acute MI. The major mediators of reperfusion injury are oxygen radicals, calcium loading, and neutrophils.23 Neutrophils are activated early during myocardial ischemia and precede the appearance of histologic tissue injury.24 Reperfusion markedly enhances the infiltration of neutrophils into the ischemic region.25 The essential initiating step involves interaction of neutrophils with vascular endothelial cells (adhesion). This is followed by activation, diapedesis, and extravascular migration into surrounding myocytes. Production of additional chemoattractants by activated neutrophils amplifies the initial inflammatory response. Neutrophil activation causes a greatly enhanced oxygen uptake by the cell, resulting in the production of large quantities of reactive oxygen species that may lead to disruption of EC and inactivate antiproteases present in the plasma.26
The term reperfusion injury was coined to describe reperfusion-related expansion or worsening of the ischemic cardiac injury as assessed by contractile performance, the arrhythmogenic threshold, conversion of reversible to irreversible myocyte injury, and microvessel dysfunction. The process of restoring blood flow to ischemic myocardium has shown utility in limiting cell death in the presence of severe ischemia. Reperfusion, however, has paradoxical effects on myocardium that can result in adverse reactions, thereby reducing its beneficial actions. During reperfusion, the myocardium is subject to abrupt biochemical and metabolic changes governed by several mediators that interact with each other in complex ways.27,28
Potential mediators of lethal reperfusion injury include oxidative stress (oxygen paradox), sudden increases in intracellular Ca2+ (calcium paradox), rapid restoration of physiologic pH (pH paradox), and inflammation. At the time of myocardial reperfusion, there is an abrupt increase in intracellular Ca2+, leading to a disturbance in the normal mechanisms that regulate Ca2+ within the cardiomyocyte, known as the calcium paradox.29 This intracellular Ca2+ overload induces death by causing hypercontracture of myofibrils and mitochondrial permeability transition pore (PTP) opening.27,29 Mitochondrial PTP is a nonselective channel located on the inner mitochondrial membrane and is a critical determinant of lethal reperfusion injury. Opening the channel, as occurs within the first few minutes after reperfusion, uncouples oxidative phosphorylation, resulting in ATP depletion and cell death.
In addition, experimental studies have shown that reperfusion of ischemic myocardium generates reactive oxygen species (oxygen paradox). This oxidative stress results in extension of the myocardial injury beyond that induced by ischemia alone. A key mechanism involves the reduction of the bioavailability of the nitric oxide, a cardioprotective signaling molecule. Nitric oxide inhibits neutrophil accumulation, inactivates superoxide radicals, and improves coronary blood flow.30 Further contributing to reperfusion injury is the rapid restoration of physiologic pH that occurs after washout of lactic acid and the activation of the sodium–hydrogen exchanger and the sodium–bicarbonate symporter.31
Finally, inflammation plays a role in reperfusion injury, primarily through release of chemoattractants, which recruit and activate neutrophils. Neutrophils release degradative enzymes and cause microvascular plugging and generation of reactive species, all of which contribute to exacerbation of the previously injured myocardium.
New experimental data into the pathogenesis of reperfusion injury have recently emerged. The RISK (reperfusion injury salvage kinase) pathway refers to a group of protein kinases that when activated during myocardial reperfusion confer cardioprotection by preventing lethal reperfusion injury.32 The RISK pathway inhibits mitochondrial PTP opening and activates anti-apoptotic pathways and may also represent a mechanism for programmed cell survival.33 Preclinical studies have shown that activation of the RISK pathway by pharmacologic agents or by mechanical interventions such as ischemic preconditioning reduce myocardial infarct size by up to 50%.34,35
Interestingly, reperfusion injury has been shown to be reduced by hypothermia when initiated late during ischemia and continuing for several hours of reperfusion, resulting in improved perfusion and reducing macroscopic zones of no-reflow and extent of necrosis in animal models.36 Although many cardioprotective strategies based on new experimental insights have been successfully applied in animal models, the beneficial effects in preclinical models have been difficult to confirm in clinical trials.
Another recent clinical study involving infusion of bone marrow–derived human mesenchymal stem cells (hMSCs) showed both safety and efficacy in patients presenting with first acute MI within 1 to 10 days. Patients treated with hMSCs who had anterior MIs had higher ejection fractions and reverse ventricle remodeling.37
Acute coronary thrombosis with or without percutaneous intervention results in the embolization of microparticles, including fragments of fibrin-platelet thrombus and necrotic core. Coronary microembolization is associated with arrhythmias, contractile dysfunction, microinfarcts, and reduced coronary reserve.38 Autopsy studies show a 13% rate of microembolization in cardiac disease, often associated with focal myocyte necrosis.39 The rate of coronary microembolization is highest in patients with documented epicardial coronary thrombosis, reaching 30% to 54%,40,41 and is even higher in those with acute infarcts (79%) in acute MI.42 Little data are available that compares acute plaque rupture with acute plaque erosion and the rate of embolization, but we have noted a higher rate of thrombotic microembolization with plaque erosion. In hearts with acute coronary thrombi, evidence of distal embolization was more frequent in erosions than ruptures (71% vs 42% P = .01).43 These emboli and microvessel obstruction have a prominent clinical role because myonecrosis is often associated with these findings.
Other potential sources of distal embolization include primary PCI, PCI of diseased (atherosclerotic) saphenous vein grafts, and thrombolysis. The reported incidence of distal embolization caused by soft friable atherosclerotic plaque and adherent thrombus after PCI of bypass grafts ranges from 2% to 42%.44-46 Angiographic evidence of distal embolization in patients presenting with acute MI undergoing primary PCI ranges from 15% to 19% (Fig. 57–5).47,48
Figure 57–5.
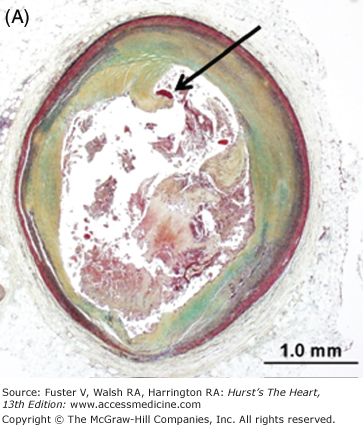
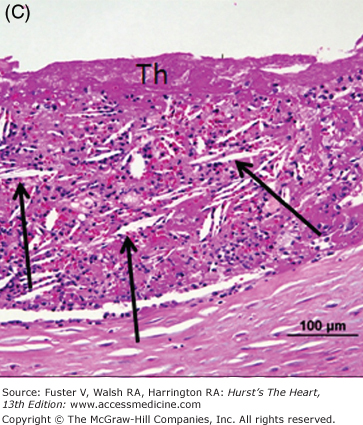
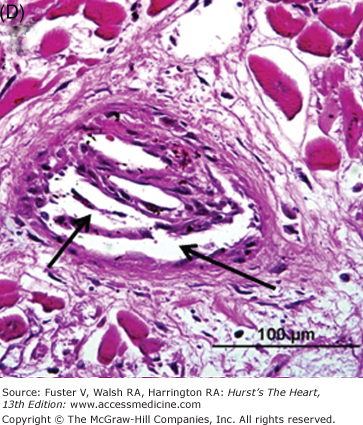
Representative micrographs of distal microembolization of plaque material from ruptured atheroma. A. Low-power (Movat pentachrome) view of rupture site (arrow) showing luminal fibrin and cholesterol clefts. B. Adjacent distal segment to rupture site demonstrates non-occlusive thrombus composed of necrotic core material. C. High-power image demonstrating cholesterol clefts (arrows) with superimposed thrombus (Th) (hematoxylin and eosin). D. High-power image of intramural coronary artery atheroembolus consisting of cholesterol clefts (arrows) and surrounded by healing myocardium in a patient who underwent percutaneous coronary intervention for acute myocardial infarction.
Ischemic preconditioning was first described by Murry et al49 in 1986, when they observed that canine myocardial infarct size was markedly reduced if it occurs after four brief episodes of 5 minutes of occlusion followed by 5 minutes of reperfusion. Preconditioning has also been observed with only one episode of brief occlusion followed by reperfusion. If the duration between ischemic preconditioning and the long duration of occlusion is extended to 24 to 96 hours, the protective effect is markedly reduced.50 The original description of preconditioning was applied only to infarct size reduction; this definition is now extended to cardiac function and arrhythmias, although the latter are not as consistent.51
The mechanisms of preconditioning are unclear, but it has been shown that preconditioning reduces the energy demand of the myocardium, both in animals and humans. Two phases of preconditioning have been described: the classical initial phase, operative for 1 to 2 hours before sustained coronary occlusion, and the delayed phase, operative 24 hours after the precondition, known as the second window of protection (SWOP).8 Classical preconditioning is associated with activation of adenosine receptors, activation of protein kinase C (PKC) coupled to G proteins, and opening of ATP-dependent potassium channels. The mechanism of SWOP is less clear and is believed to involve a kinase cascade, including mitogen-activated protein kinases and nuclear factor B, which increase levels of superoxide dismutase, nitric oxide synthase, cyclooxygenase-2 (COX-2), and heat shock proteins, thereby creating a protective milieu for the cardiomyocyte.
Clinically, Yellon et al52 showed that intermittent aortic cross-clamping could precondition the human left ventricle during coronary artery bypass surgery, resulting in preservation of ATP levels. Other observations that confirm the existence of preconditioning in patients have been observed in patients undergoing percutaneous transluminal coronary angioplasty (PTCA). Repeated balloon inflations of 60 to 90 seconds are associated with decreasing chest pain, a reduction in ST-segment elevation, and a decrease in lactate production with subsequent inflations; the foregoing phenomenon are observed irrespective of the presence or absence of collaterals.53 In the Thrombolysis in Myocardial Infarction (TIMI)-9 study, the timing of angina in relationship to MI was studied, and only patients with angina within 24 hours of infarction showed smaller infarct size and better clinical outcome.54
The mediators of preconditioning are believed to involve the KATP channel and specific isoforms of PKC. The protective effect of temporary ischemia can be blocked by pretreatment of the myocardium with inhibitors of KATP channel, such as glibenclamide and 5-hydrocydeconate (5HD).55,56 Similarly, inhibitors of PKC and tyrosine kinase, but not PKC alone, will prevent ischemic preconditioning. Also similarly, agonists of adenosine (A1 receptor) will pharmacologically precondition the heart against ischemia.57
Left ventricular systolic dysfunction caused by ischemia may arise in dead myocardium or from hypocontractile areas of viable myocardium. In the early 1980s, Rahimtoola et al58 found significant improvement in left ventricular function after coronary revascularization in a subset of patients with depressed ventricular performance. They postulated that the mechanism of poor myocardial contractility was chronic ischemia, which could be improved by revascularization. The premise behind this rationale was dependent on the surviving myocardium being in a functional, albeit depressed, state, suggesting that the myocardium may adapt to chronic ischemia by decreasing its contractility but preserving viability.59 Reversibly, dysfunctional tissue is commonly referred to as hibernating myocardium.60 Sheiban et al61 demonstrated that 5 to 7 minutes of angioplasty balloon inflations in the coronary arteries of patients undergoing interventional procedures, followed by tracking of the resolution of the regional wall-motion abnormalities over the next 5 days, showed persistence of regional wall motion abnormalities for up to 36 hours. Similarly, return of left ventricular function has been studied after acute MI. Delayed recovery of wall motion was observed in the infarct region, with a positive change in wall motion from 0.2 at 3 days to 1.0 at 6 months in patients with reperfusion but not in those without reperfusion, as measured by the centerline method.62
Clinical functional techniques such as stress echocardiography and cardiac magnetic resonance are more specific but less sensitive than nuclear modalities, which assess perfusion and metabolic activity, in the detection of hibernating myocardium.60,63 The concept of myocardial hibernation was initially based on clinical observation. Subsequently, there have been a number of experimental studies to support the concept that ischemia is not the result of simple inadequacy of blood flow for myocardial contraction but that there is a stepwise decrease in function based on incremental decrease in oxygen-supplying perfusion (so-called perfusion–contraction matching). Evidence suggests that repeated episodes of ischemia–reperfusion may result in a state of chronic hibernation, with alterations in the flow–function relationship and decreased oxygen demand. Chronically hibernating myocardium demonstrates alterations in adrenergic control and calcium responsiveness. Substances that are upregulated in chronic hibernating myocardium include heat shock protein, hypoxia-inducible factor, inducible nitric oxide synthase, COX-2, and monocyte chemotactic protein. Because some of these pathways are involved in preconditioning, a relationship between cardiac hibernation and preconditioning is postulated.
Morphologically, hibernating myocytes show loss of contractile elements, especially in the perinuclear region, and occasionally throughout the cytoplasm. The space left by the dissolution of the myofibrils is occupied by glycogen, as evidenced by the strong positivity for the periodic acid-Schiff reagent. Ultrastructurally, there is depletion of sarcomeres, most pronounced in the perinuclear region, with increased glycogen. The nuclei are enlarged, with a tortuous nuclear membrane and evenly distributed heterochromatin. The mitochondria are elongated, shrunken, and osmiophilic.64 The interstitium shows an increase in connective tissue. Increased numbers of apoptotic myocytes, using the technique of DNA nick-end labeling, ,65 as well as an increase in autophagic and oncotic cell death, have been demonstrated.10,66
The composition and distribution of sarcomeric, cytoskeletal, and membrane-associated proteins is significantly altered in chronic myocardial hibernation.67,68 There is a disorderly increase in cytoskeletal desmin, tubulin, and vinculin, with a decrease in contractile proteins myosin, titin, and actinin. More recently, decreased connexin 43, a membrane transport protein, has been associated with reduced gap junction size and a proposed propensity for arrhythmias in the hibernating state.69
Plaque Morphology and Site of Thrombus in Acute Myocardial Infarction
The vast majority of MIs occur in patients with coronary atherosclerosis, with more than 90% associated with superimposed luminal thrombi. Arbustini et al70 found coronary thrombi in 98% of patients dying with clinically documented acute MI, and of those thrombi, 75% were caused by plaque rupture and 25% by plaque erosion. There are gender differences in the causation of coronary thrombi leading to acute MI: Arbustini et al70 showed that 37% of thrombi in women were erosion compared with only 18% in men. Whereas the majority of acute thrombi (<1 day) have been observed in plaque rupture, the majority of thrombi in plaque erosion are organizing (>1 day)71 (Fig. 57–6). Kramer et al71a have reported their findings in thrombi retrieved from patients presenting with ST-segment elevation MI (STEMI) during percutaneous coronary angioplasty intervention and demonstrate that more than 50% of thrombi show organization of more than 1 day’s duration. Furthermore, the age of the thrombus, in addition to other risk factors, was an independent predictor of long-term mortality, especially within the first 2 weeks as well as at 4 years after PCI.
Figure 57–6.
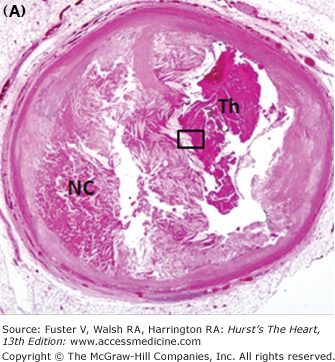
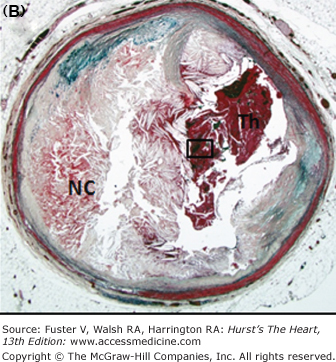

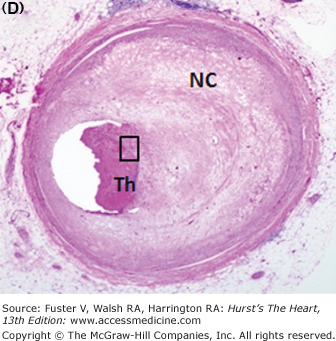

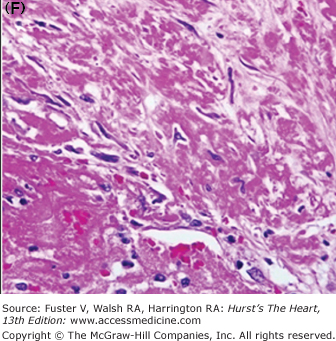
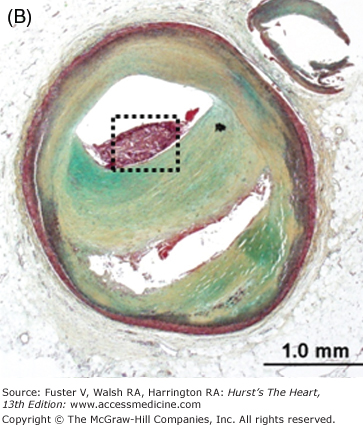
Morphology of coronary thrombi associated with plaque rupture and erosion. A. and B. Low-power views of a human ruptured coronary lesion showing a relatively large necrotic core (NC) and superimposed thrombus (Th) (early, nonhealing, <1 day in age). C. Higher magnification of thrombus area referenced by the insert in A showing platelets and fibrin and focal collections of neutrophils. D. and E. Low-power views of a human ruptured coronary lesion showing a macrophage-rich early fibroatheroma with coronary plaque erosion with a superimposed healing thrombus (>7 days of age). F. Higher magnification of healing thrombus composed of layers of smooth muscle cells and proteoglycan-collagen matrix. Parts A to D, X 20 magnification, hematoxylin and eosin (H&E) staining; parts B and E, X20 magnification, Movat pentachrome staining; parts C and F, X400 magnification, H&E staining.
Although an individual severe stenosis is more likely to become occluded by a thrombus than a lesion with less severe stenosis, the less severely narrowed plaques give rise to more occlusions because there are many more sites that are mild to moderately narrowed.72 We have observed that the mean percent stenosis underlying coronary plaque erosion is 70% versus 80% at the site of plaque rupture; however, 82% of fatal plaque erosions result in total occlusions compared with only 57% of plaque ruptures.73 The majority of thin cap fibroatheromas (TCFAs), acute and healed ruptures, and lesions with fibroatheromas occur predominantly in the proximal portion of the three major coronary arteries, and about 50% arise in the midportion of these arteries. By far the proximal portion of the left anterior descending coronary artery (LAD) is the most frequent location, with sites in the proximal right and left circumflex contrary arteries about half as common. The culprit coronary artery of infarction at autopsy most frequent is the LAD (~50%) followed by the right coronary artery (30%-45%) and then the left circumflex artery (15%-20%).71 No thrombi are found in fewer than 5% of acute MIs.
Pathology of Acute Myocardial Infarction
The earliest change that can be grossly discerned in the evolution of acute MI—pallor of the myocardium—occurs 12 hours or later after the onset of irreversible ischemia. The gross detection of infarction can be enhanced by the use of tetrazolium salt solutions, which form a colored precipitate on gross section of fresh heart tissue in the presence of dehydrogenase-mediated activity. The tetrazolium salts (nitroblue tetrazolium [NBT] and 2,3,5-triphenyltetrazolium chloride [TTC]) are dyes that are sensitive to the presence of tissue dehydrogenase enzyme activity, which is depleted in the infarcted myocardium. It has been shown that myocardial infarct can be detected by NBT as early as 2 to 3 hours in dogs and in a little less time in pigs because of poor collaterals.74 Red color (TTC) or blue color (NBT) only forms in the normal noninfarcted myocardium, thus revealing the pale, nonstained, infarcted region (Fig. 57–7). In humans, the necrotic myocardium can be detected within 2 to 3 hours after infarct by immersion of the fresh heart slices in a solution of TTC or NBT. TTC staining has a diagnostic sensitivity of 77% and a specificity of 93% compared with routine histology, with predictive values of positive and negative test of 81% and 91%, respectively.75
Figure 57–7.
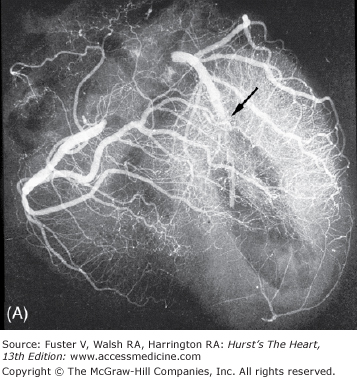
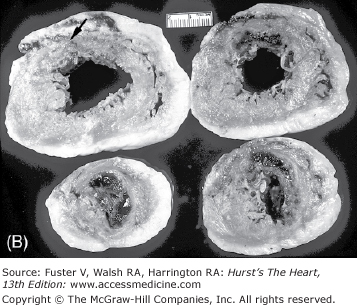
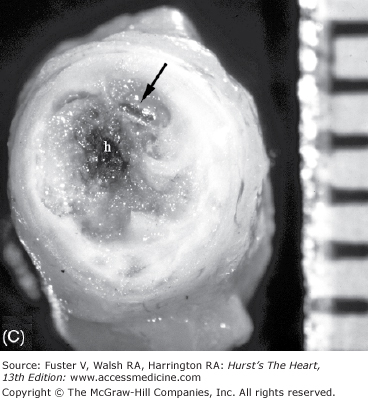
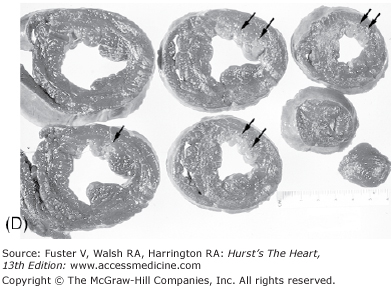
Regional distribution of vascular supply to the ventricles with right coronary artery dominance. A. Postmortem angiogram of the heart in a patient with acute myocardial infarction with total occlusion (arrow) of the proximal left anterior descending coronary artery (LAD) in a 65-year-old woman who presented with persistent chest pain of 6 hours’ duration. B. At autopsy, she had a hemopericardium with rupture site (arrow) identified on the anterior wall of the left ventricle. Note extensive hemorrhagic (h) transmural infarction involving the anterior wall of the left ventricle near the base of the heart (upper slices) and extending into the septum in the mid and apical slices (lower slices). C. Gross photography of the LAD showing hemorrhage into the necrotic core and more than 90% luminal narrowing; barium is seen within the lumen (arrow). D. Dog heart slices after 15 minutes of incubation in 2% triphenyltetrazolium chloride (TTC) at 98.6°F (37°C). The animal had undergone 60 minutes of LAD occlusion distal to the first diagonal branch followed by reperfusion and was sacrificed at 24 hours. Injecting monastery dye after reocclusion of the LAD and just before sacrifice identified the myocardium at risk of infarction. The heart was sliced and then immersed in TTC. The viable myocardium at risk stains red, the area not at risk is bluered, and the infarcted region is creamy white (arrows
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


