Chapter 113 Patent Ductus Arteriosus, Coarctation of the Aorta, and Vascular Rings
PATENT DUCTUS ARTERIOSUS
Patent ductus arteriosus was first described by Galen in the 3rd century and subsequently by Leonardo Botallo in the 15th century. The first surgical closure of a PDA was reported by Drs. Gross and Hubbard in 1939. The prevalence of PDA in neonates at several days of age varies from 20% to 80% and varies inversely with gestational age and birth weight.1 It is frequently associated with other heart defects, ranging in severity from patent foramen ovale to complex congenital malformations. Several genetic defects have been implicated in the development of PDA, including genes that encode prostaglandin receptors and regulators of smooth muscle cell contraction.2–4
Pathophysiology and Natural History
In utero, the ductus arteriosus shunts mostly right to left because of the high resistance of the pulmonary circulation. Shortly after birth, expansion of the lungs results in a decline in the pulmonary vascular resistance and increased pulmonary blood flow and arterial oxygen tension. Ductal closure usually occurs within the first several hours after birth and is thought to be mediated by loss of the placental source of prostaglandins, increased degradation of prostaglandins in the lungs, and increased arterial oxygen tension, which stimulate constriction of sensitive smooth muscle cells in the wall of the ductus. The prostaglandin-mediated mechanism is most active in the ductus of premature infants, whereas oxygen tension primarily promotes ductal closure in the term infant, probably because of differences in cyclooxygenase (COX) isoforms present in the ductal tissue at various stages of gestation.5 Thus the PDA of a term infant is generally unresponsive to indomethacin treatment, whereas the PDA of a premature infant frequently responds.
Spontaneous closure of the ductus within 4 days occurs in 90% to 95% of full-term infants and 80% to 90% of premature infants at 30 to 37 weeks’ gestational age.6 The rate of spontaneous closure is inversely proportional to birthweight, with only 50% spontaneous closure in extremely low birthweight infants (500 to 999 g).7 In premature infants, PDA increases the risk of prolonged ventilation and oxygen requirements, pulmonary hemorrhage,8 and bronchopulmonary dysplasia (BPD).9–11 The diastolic steal is associated with renal hypoperfusion,12 intestinal ischemia, necrotizing enterocolitis (NEC), reduced middle cerebral artery blood flow velocity,13–16 and increased risk of intraventricular hemorrhage (IVH).17,18 The long-term risks of PDA in term infants include development of endocarditis, of congestive heart failure, and, eventually, of irreversible pulmonary vascular obstructive disease. Estimated mortality without intervention is 60% by 60 years of age.19
Diagnosis
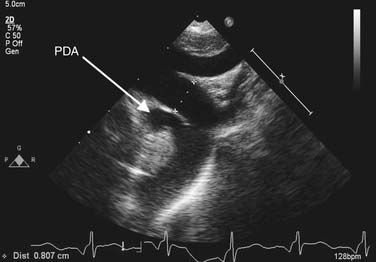
Chest radiography may show cardiomegaly, signs of pulmonary congestion, and pulmonary edema. In older patients, calcification in the wall of the ductus or enlarged pulmonary arteries, characteristic of pulmonary hypertension, may be noted. Transthoracic echocardiography is used to make the diagnosis (Fig. 113-1). The presence of a left-sided aortic arch must be confirmed if surgical therapy is contemplated, as this influences the choice of incision. A commonly associated finding in premature infants is the presence of a patent foramen ovale, which spontaneously closes over time. However, its presence becomes significant in an adolescent or adult with a calcified ductus who requires cardiopulmonary bypass, as it would alter the cannulation strategy at the time of ligation. Continuous left-to-right shunting through the ductus is expected, and the presence of right-to-left shunting or bidirectional shunting should raise suspicion for pulmonary hypertension. In adults, echocardiographic windows may be suboptimal, and CT or MR angiography may be necessary to establish the diagnosis. Diagnostic cardiac catheterization is necessary only if pulmonary hypertension is suspected.
Treatment
Medical
Medical treatment of PDA in premature neonates can be classified as prophylactic or symptomatic. Prophylactic medical treatment with COX inhibitors within the first 24 hours, even in asymptomatic patients, improves the rate of ductal closure and the incidence of IVH, and it decreases the need for surgical closure, but it does not improve mortality or incidence of NEC. Early treatment of a symptomatic PDA (i.e., when clinical signs first appear) has been shown to decrease the incidence of chronic lung disease and NEC as well as the duration of mechanical ventilation when compared with late symptomatic treatment (i.e., after signs of congestive cardiac failure).20 The COX inhibitors indomethacin and ibuprofen are equallyt effective at producing ductal closure. Closure rate between 60% and 80% is achieved with one course of indomethacin, but the success rate of each subsequent course is 40%.21,22 COX inhibitors are ineffective at producing ductal closure in term infants because of the lack of prostaglandin-responsive contractile smooth muscle in their ductal tissue. Complications due to indomethacin include renal impairment, intestinal perforation, and NEC.23
Surgical
Premature Infants
In premature infants, surgical therapy is generally reserved for failed medical therapy in symptomatic patients and for those who develop contraindications to COX inhibitor therapy—NEC, renal dysfunction, and IVH. This strategy of reserving surgery for failure of medical therapy may lead to a higher incidence of NEC when compared with early surgery, although no difference in mortality has been demonstrated.24–27 Early surgical duct closure allows early institution of full oral feeding, but this does not necessarily translate into shorter hospital stay.28
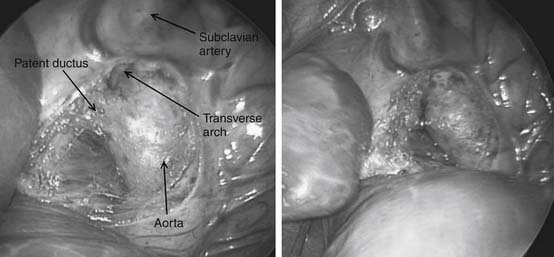
Preoperative evaluation is critical, particularly in the premature infant who is likely to have multiple comorbidities. Emphasis should be on medical stabilization, exclusion of active infections, and appropriate treatment of preexisting infections before surgery. Surgery for premature infants can be performed either in the intensive care unit or in the operating theater. Surgical ductal ligation is generally performed via a left thoracotomy, although the video-assisted thoracoscopic surgery (VATS) approach has been described, and the PDA is occluded by placement of an external clip (Fig. 113-2).
Full-Term Infants, Children, and Adults
Surgical or transcatheter closure is the mainstay of therapy for full-term infants, children, and adults, as medical therapy is usually ineffective. Ductal closure is indicated in both symptomatic and asymptomatic patients to reduce the risks of endocarditis and pulmonary hypertension. Asymptomatic infants may undergo elective closure between 1 and 2 years of age to facilitate VATS or transcatheter closure, whereas symptomatic infants should undergo prompt evaluation and closure. Options for closure in infants include thoracotomy or VATS. Ligation may be performed with sutures or clips. To avoid the risk of recanalization, some advocate ductal division in addition to ligation in children and adults.29
Children and adults are candidates for closure via the transcatheter, thoracotomy, VATS, or sternotomy approach. The optimal approach depends on the morphology of the ductus. Short length and large diameter of the PDA increase the risk of device embolization during transcatheter closure, and bleeding during VATS closure.30 Extensive ductal calcification increases the risk of bleeding during closure by VATS or thoracotomy. In these patients, median sternotomy, cardiopulmonary bypass, and closure through a pulmonary arteriotomy may be performed. The presence of cyanosis and bidirectional shunting through the PDA, demonstrated by echocardiography, should prompt preoperative cardiac catheterization to measure pulmonary vascular resistance. If elevated pulmonary vascular resistance is encountered that is unresponsive to oxygen or nitric oxide, closure is contraindicated.
Prognosis
Operative mortality related to PDA ligation in full-term infants and children is less than 1%.29 Premature infants with multiple comorbidities may have hospital mortality as high as 20%. Complications of surgical ligation include left recurrent laryngeal nerve injury, bleeding, postoperative chylothorax, and development of coarctation. Asymptomatic recurrent laryngeal nerve injury can be detected in up to 7% of patients by systematic endoscopy, and the major risk factor for this injury is birth weight less than 1 kg.31 Symptomatic nerve injury (e.g., feeding difficulty, aspiration) is rare.
COARCTATION OF THE AORTA
Coarctation of the aorta comprises 4% to 8% of cases of congenital heart disease, with an incidence in the general population estimated at 0.2 per 1000 live births. The anomaly occurs twice as often in males as in females. It can occur in association with a variety of other heart defects—50% of patients with coarctation of the aorta have bicuspid aortic valve. A ventricular septal defect (VSD), seen in 30% to 60% of patients, may be associated with posterior malalignment of the conal septum leading to left ventricular outflow tract obstruction. A thorough examination of the intracardiac anatomy is necessary, because associated hypoplasia of left heart structures is relatively common. Complex congenital heart disease associated with hypoplasia of the systemic ventricle (hypoplastic left heart syndrome, right dominant atrioventricular canal, transposition of the great arteries with hypoplastic right ventricle) may also have associated coarctation, which would have an impact on management. The incidence of coarctation in patients with right aortic arch is unknown, but it has been estimated to be approximately 4%.32
Anatomy and Pathophysiology
The etiology of coarctation with hypoplasia of the distal transverse arch is not well defined, but its association with hypoplasia of left heart structures suggests common genetic, hemodynamic, or environmental mechanisms.33 Discrete coarctation of the aorta at the isthmus probably results from abnormal infiltration of ductal tissue onto the juxtaductal aorta.34 The contractile ductal tissue in the aorta constricts synchronously with ductal closure, resulting in luminal narrowing of the aorta and development of a posterior shelf. It is not uncommon for the aorta to appear to be of adequate size in the presence of a patent ductus arteriosus, yet manifest coarctation after ductal closure.
Natural History
Collateral vessels become prominent as a result of increased flow in the intercostal, internal mammary, and scapular blood vessels. The increased flow in these vessels can be sufficient to reduce the blood pressure gradient between the upper and lower extremities at rest. With exercise, however, the gradient may rise significantly because of the elevated vascular impedence. Upregulation of circulating catecholamines and the renin-angiotensin system leads to systemic hypertension. Left ventricular hypertrophy is a result of elevated vascular resistance. Untreated coarctation is associated with a substantially diminished long-term survival, with 75% mortality by age 50.35 Death in these patients is usually the result of systemic effects of hypertension, including heart failure, intracranial hemorrhage, coronary artery disease, or aortic rupture or dissection.
Diagnosis
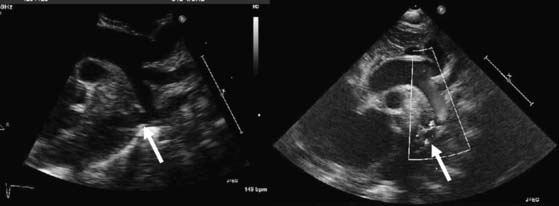
The segment of coarctation can usually be visualized easily in neonates and children by two-dimensional echocardiography (Fig. 113-3). Normalized measurements of the transverse aorta, aortic isthmus, and descending aorta mustt be obtained. In neonates, a Z-score of less than −2 indicates significant narrowing. In older children and adults, a greater than 50% decrease in diameter at the aortic isthmus should be considered severe coarctation. Associated VSD, aortic and mitral valvular abnormalities, and ventricular hypoplasia must be ruled out. Pulsed and continuous wave Doppler can be used to estimate the pressure gradient directly at the area of coarctation. In the neonate, the pressure gradient measured by echocardiography may be confounded by the presence of a PDA or severe ventricular dysfunction. Similarly, in the older child or adult, the pressure gradient may underestimate the severity of the narrowing when significant collaterals are present. A peak gradient of greater than 20 mm Hg, especially if accompanied by continuous forward flow during diastole in the descending or abdominal aorta, suggests significant aortic coarctation.
Treatment
Surgical Repair
For patients with left aortic arch, the approach is via a left thoracotomy. Primary treatment for neonates and infants is extended end-to-end anastomosis (Fig. 113-4). In this technique, the descending aorta, transverse arch, and branch vessels are mobilized. The descending aorta and distal transverse arch are clamp occluded, and the ductus arteriosus is divided. The coarctation segment is resected, and the undersurface of the arch is opened as far proximal as the takeoff of the left carotid artery, and the anastomosis between the descending aorta and the aortic arch is performed. Patch angioplasty or interposition grafts are typically necessary for adults and adolescents, in whom limited aortic mobility prevents a tension-free aortic anastomosis. Subclavian flap angioplasty involves division of the left subclavian artery and use of the proximal portion for aortic reconstruction. The flap is placed on the aorta distal to the subclavian artery for traditional coarctation repair, whereas a reverse flap is used to augment the transverse arch when hypoplasia between the left carotid and subclavian artery is encountered.
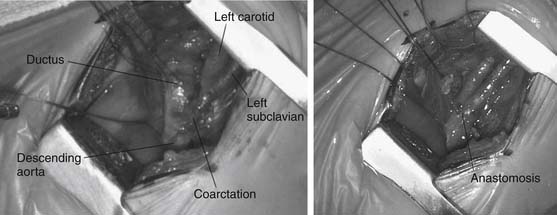
Figure 113–4 Intraoperative appearance of coarctation segment (left) and extended end-to-end anastomosis (right).
Coarctation in patients with right aortic arch is approached by a right thoracotomy, and treated by patch angioplasty rather than extended end to end anastomosis.32 Although repair of coarctation through a left thoracotomy does not require circulatory support, partial bypass may be helpful in adolescents and adults in whom collateral vessels are underdeveloped or have been surgically interrupted. Partial left heart bypass is instituted by providing venous drainage (left atrial appendage or pulmonary vein) and arterial cannulation (femoral artery or descending aorta). Native heart ejections are maintained to provide antegrade blood flow to the upper body during the crossclamp period. In rare circumstances, ascending to descending aortic grafts are used for repair of native or recurrent coarctation associated with significant cardiac disease that requires concomitant surgical management (e.g., coronary artery bypass grafting, aortic valve replacement). Access to the descending aorta can be obtained by incising the posterior pericardium and dissecting the supradiaphragmatic aorta. Ascending to abdominal aorta bypass avoids the risk of paraplegia associated with left thoracotomy repair of recoarctation in adults.36
Coarctation with Ventricular Septal Defect
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


