Initiation
SIRS is incited by a number of factors, including the interaction of blood with the foreign surfaces of the CPB circuit (i.e. contact activation), altered blood flow patterns (i.e. non-pulsatile flow), ischemia-reperfusion injury, and endotoxins.
Initiation results from a multitude of cellular-mediated processes including:
2. Complement activation
3. Kinin system activation
4. Fibrinolytic system activation
5. Synthesis of cytokines
6. Neutrophil activation
1. Endothelial activation
The endothelium lines the interior surface of blood and lymphatic vessels. Physiologically, the endothelium plays a dynamic protective role by: (1) selectively controlling passage of material and white blood cells into and out of the circulation, (2) inhibiting clot formation by secreting anticoagulants such as tissue plasminogen activator (tPA), thrombomodulin, and heparin-like substances, and (3) causing local vasodilation through nitric oxide and prostacyclin. Activation or disruption of the endothelial layer leads to a cascade of events including recruitment and release of immune cells, and release of inflammatory cytokines, procoagulant factors, and vasoactive agents. This results in further direct and indirect injury.
2. Complement activation
An essential component of the immune system, the complement system consists of over 30 plasma proteins that are involved in chemoattraction, activation, opsonization, and cell lysis. Activation of the system can occur via either the “classical” or “alternative” pathways. The two complement pathways are shown in Figure 11.2. The classical pathway involves the activation of C1 by an antibody–antigen complex. The alternative pathway does not require an antibody for its activation: C3 fragments free-floating in serum attach directly to antigens, endotoxins, or foreign surfaces (otherwise known as contact activation). The common step linking both pathways is the cleavage of C3. Cleavage of C3 to its activated form C3a stimulates the release of histamine and other inflammatory mediators from mast cells, eosinophils, and basophils. This results in smooth muscle constriction and an increase in vascular permeability. C5a is a potent chemotactic factor for neutrophils as it promotes their aggregation, adhesion, and activation. C3b and C5b interact on cell membranes with components C6–C9 to form a “membrane attack complex,” which activates platelets and “punches” holes in cell membranes.
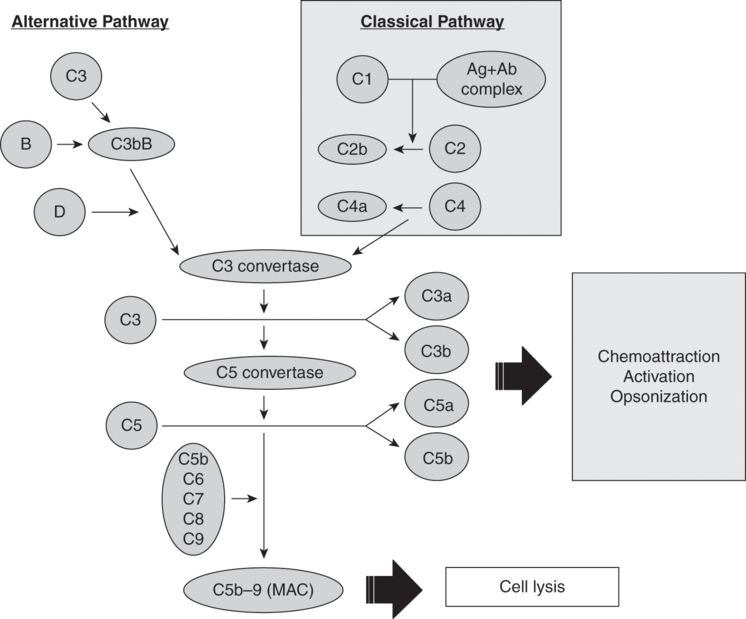
This process is rapid: plasma levels of activated complement factors rise within 2 minutes of the onset of bypass, and a second rise can be detected after release of the aortic cross-clamp and re-warming. Levels decline postoperatively and generally return to normal 18–48 hours postoperatively.
3. Kinin system activation
Initiated by contact with anionic surfaces, the initial step of the kinin system is activation of factor XII to factor XIIa (see Figure 11.3). In the presence of high-molecular-weight kininogen (HMWK), factor XIIa activates plasma prekallikrein to kallikrein. Kallikrein has five main actions:
i. accelerates and activates plasma prekallikrein via a positive feedback loop;
ii. cleaves HMWK to release bradykinin (a potent vasodilator that increases vascular permeability);
iv. activates the fibrinolytic pathway by stimulating tissue plasminogen activator; and
v. activates the sympathoadrenal system.
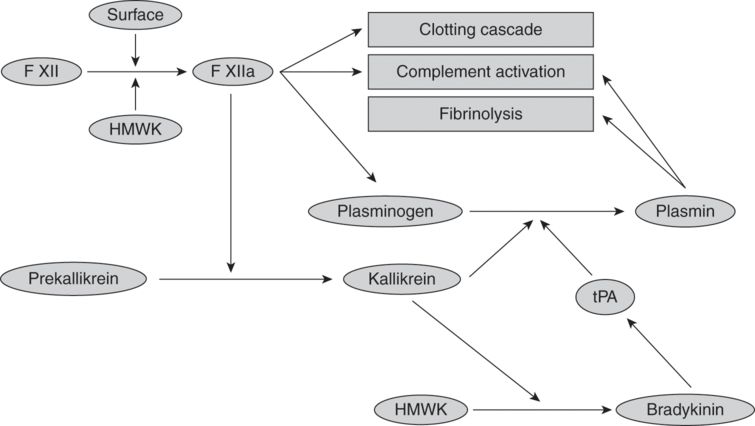
Diagram of kinin, clotting, and fibrinolytic system. Abbreviations: tPA = tissue plasminogen activator; HMWK = high-molecular-weight kininogen.
4. Fibrinolytic system activation
Exposure of blood to the artificial surface of the CPB circuit, temperature changes, medications, mechanical trauma, or blood products trigger fibrin clot formation, resulting in activation of the fibrinolytic system. The fibrin clots are continuously proteolytically digested into fibrin degradation products (FDPs) by plasmin (see Figure 11.3). Plasmin production is upregulated via two pathways:
i. Bradykinin upregulates tissue plasminogen activator (tPA), which in turn converts plasminogen to plasmin. tPA levels peak with 30 minutes of CPB and return to baseline within 24 hours.
Intravascular fibrin clots lead to impaired microcirculation and hypoxic cellular damage. FDPs compete with thrombin and slow down clotting by inhibiting the conversion of fibrinogen to fibrin. The net effect is endothelial and platelet dysfunction.
5. Synthesis of cytokines
Cytokines are soluble proteins released by immune cells and endothelial cells in response to local injury and play a critical role in the pathophysiology of inflammation. Cytokines are important messengers in the immune system: they attract immune cells to areas of injury and enhance their function. In addition, they stimulate further expression of procoagulant and fibrinolytic enzymes. Proinflammatory cytokines tumor necrosis factor (TNF), interleukin-1 (IL-1), IL-6, IL-8, and IL10 are all elevated during CPB. Excessive levels of cytokines lead to uncontrolled systemic inflammation causing tissue damage.
6. Neutrophil activation
Recruitment and activation of neutrophils occurs in response to local endothelial damage and cytokine release, resulting in leukocyte adhesion. Leukocytes cause local damage via three main mechanisms:
i. aggregation with platelets to cause local microvascular occlusion and ischemia,
ii. release of proinflammatory cytokines,
Cellular injury
Described throughout the previous text, the mechanisms for cellular and organ injury are:
1. Microvascular occlusion
2. Vasoconstriction
3. Fibrinolysis
4. Thrombosis
Ischemia-reperfusion injury
Ischemia-reperfusion injury is a term used to describe the cellular injury that occurs on resumption of normal perfusion to an organ after a period of relative or complete ischemia. During ischemia, there are a multitude of cellular changes including:
2. increase in intracellular calcium (Ca2+),
3. increase in hypoxanthine,
4. decrease in adenosine triphosphate (ATP),
5. cellular acidosis, and
6. edema.
Upon reperfusion, a cascade of mechanisms occurs that may contribute to further cell death due to alteration in calcium handling, oxygen free radical production, and hypoxanthine accumulation.
Altered calcium handling
During ischemia, there is an increase in intracellular Ca2+, primarily due to failure of ATP-dependent cellular calcium pumps. On reperfusion, there is an increase in both intracellular and mitochondrial Ca2+ secondary to entry via L-type Ca2+ channels and alterations of sarcoplasmic reticulum Ca2+ cycling. Increased Ca2+ can lead to cell death by hypercontracture (via interaction of actin and myosin) and opening of mitochondrial permeability transition (MPT) pores. Although the exact mechanism is unknown, induction of MPT pores causes an increase in mitochondrial permeability, disrupting the electrochemical gradient essential for ATP production. This process ultimately leads to cell death.
Oxygen free radical production
Low levels of oxygen free radicals and oxidants are normal and play essential roles in cellular homeostasis. However, on reperfusion of ischemic tissue, large quantities of oxygen free radicals are generated, overwhelming the endogenous scavenging systems. Oxygen free radicals have several detrimental effects including:
i. inhibition of ATP-dependent Ca2+ pumps causing increased intracellular Ca2+;
ii. lipid peroxidation resulting in cell membrane breakdown and edema; and
iii. promotion of neutrophil transmigration and activation.
Neutrophils are the final mediators of ischemia-reperfusion injury by the production of toxic chemicals generated during the metabolism of oxygen, and by the secretion of proteolytic enzymes released from granules stored in their cytoplasm.
Hypoxanthine accumulation
During ischemia, energy generation using high-energy phosphates (ATP) creates the metabolite hypoxanthine. Normally, hypoxanthine is metabolized by the enzyme xanthine dehydrogenase. However, following rapid reperfusion with increased oxygen availability, xanthine dehydrogenase is converted to xanthine oxidase. As a result, molecular oxygen is converted into highly reactive superoxide and hydroxyl radicals. Furthermore, nitric oxide produced during reperfusion reacts with superoxide to produce the potent free radical peroxynitrite. These free radicals attack and damage cellular components.
Endotoxins
Another well-described phenomenon associated with SIRS is a rise in plasma endotoxin levels during CPB. Endotoxins are lipopolysaccharides derived from the cell membranes of gram-negative bacteria. Endotoxins are produced by intestinal flora and are normally confined to the intestinal lumen by a barrier of endothelial cells. The most prominent hypothesis is that during CPB, hypoperfusion of the intestines leads to increased endothelial permeability, allowing translocation of endotoxins into the portal circulation. Lipopolysaccharides invoke potent inflammatory and immunological reactions, including macrophage activation and cytokine generation (TNF, IL-6, and IL-8).
Embolism
Intraoperative emboli during cardiac surgery can arise from many sources. With aortic manipulation (e.g. cannulation and cross-clamping), atheromatous plaques can become dislodged and scatter systemically. Interaction between the CPB circuit and blood, stagnation within the CPB circuit, direct tissue damage (i.e. to bone marrow), and introduction of homologous transfused blood can all result in microemboli. These emboli may be composed of a variety of biological products or aggregates including fibrin/fibrinogen, platelets, lipids, and protein. Gas emboli can originate from the CPB apparatus or be entrapped directly from the surgical field. Lastly, emboli can be composed of foreign material, such as plastic from the CPB circuit tubing, components of surgical items (e.g. fibers, plastics, metals), and bone wax. Emboli cause acute disruption of blood flow, leading to upstream ischemia and eventual cell death. The extent of damage is contingent on the territory supplied by the affected vessels and the duration of ischemia.
Non-pulsatile flow
Most cardiac centers employ non-pulsatile roller or centrifugal pumps to drive circulation during CPB. Though the exact mechanism is not fully understood, it is believed that non-pulsatile laminar flow, as opposed to physiological pulsatile flow, causes deterioration of microcirculatory perfusion. The net result of this derangement is an increase in endotoxins, increase in endogenous vasoconstrictors, and platelet activation leading to local cellular damage. On the other hand, roller pumps designed to rotate at varying speeds can achieve pulsatile flow in an appropriately configured CPB circuit. It has the advantage of delivering more energy to the microcirculation. Theoretically this should result in preservation of the microcirculation, improvement of the blood flow to the vital organs, and attenuation of the systemic inflammatory response. Despite these potential advantages, the use of pulsatile flow during CPB is still controversial among surgeons because of the absence of clinical data showing any clinically significant benefits and the perception that pulsatile CPB is more technically challenging to provide.
Hemodilutional anemia
Hemodilutional anemia is an inevitable consequence of CPB, given the current practice of priming the CPB circuit with non-blood components. There are advantages and disadvantages to hemodilution. The main advantage is reduced blood viscosity improving microvascular flow. The main disadvantage of excessive hemodilution is increased risk of compromising oxygen delivery at the tissue level, leading to ischemic organ injury.
Temperature
Thermoregulation is an essential homeostatic process: regulation of core body temperature at 37°C is one of the functions of the hypothalamus, ensuring optimal physiological conditions. During CPB, the physiological balance is altered, leading to a multitude of injuries. Hypothermic strategies are often employed during CPB to provide organ protection. The application of hypothermia (< 34°C) during CPB has been in practice during coronary artery bypass grafting and valvular heart disease for over half a century. The primary mechanism is a reduction in metabolic rate and oxygen consumption, but other possible benefits include attenuation of excitatory neurotransmitters (particularly glutamate), preservation of phosphate stores, and reduction in cytokine release and inflammation.
Extremes of temperature and rapid re-warming can nullify the beneficial effects of mild hypothermia. The main detriments of very low temperatures are decreased macro- and microvascular blood flow and increased blood viscosity. In addition, hypothermia can lead to leukocyte activation, endotoxin release, and production of TNF. All the aforementioned mechanisms can lead to perioperative organ damage. Both rapid re-warming and inadvertent hyperthermia during CPB can cause organ damage. The mechanism of this damage includes production of free radicals, intracellular acidosis, increase in excitatory amino acid neurotransmitters, and hyperemia. Despite the theoretical advantages of hypothermia during CPB, studies have failed to show a clinical advantage of hypothermia in respect to cerebral protection during CPB. In fact, no difference between CPB utilizing hypothermia or normothermia (> 34°C), with respect to mortality rate, stroke, cognitive decline, myocardial infarction, use of inotropic support or intra-aortic balloon pump, infections, atrial fibrillation or acute kidney injury has been conclusively demonstrated. In contrast, hypothermia was associated with coagulopathy, increased risk of transfusions, impaired drug metabolism, and longer periods of bypass time. The release of markers of myocardial injury during CPB seems to be independent of the temperature used.
Alterations in organ perfusion
The transition from physiological circulation to CPB represents a major change to the homeostasis of the body. This is secondary to changes in the type of circulation from pulsatile to non-pulsatile flow and in the temperature of the body from normothermia to hypothermic circulation. This transition leads to alterations in distribution of blood flow and oxygen delivery to the organs (see Figure 11.4a). Furthermore, tissue oxygen delivery is influenced to a large extent by CPB flow rate (see Figure 11.4b). Organ dysfunction may, thus, be in part attributed to these changes in the regional distribution of blood flow and the dependence of oxygen delivery on the maintenance of adequate CPB flow rates. Also, the initiation of the SIRS contributes to many of these complications. Table 11.1 summarizes a list of complications post-cardiac surgery. Cerebral and renal complications are discussed in further detail in Chapters 12 and 13 respectively.
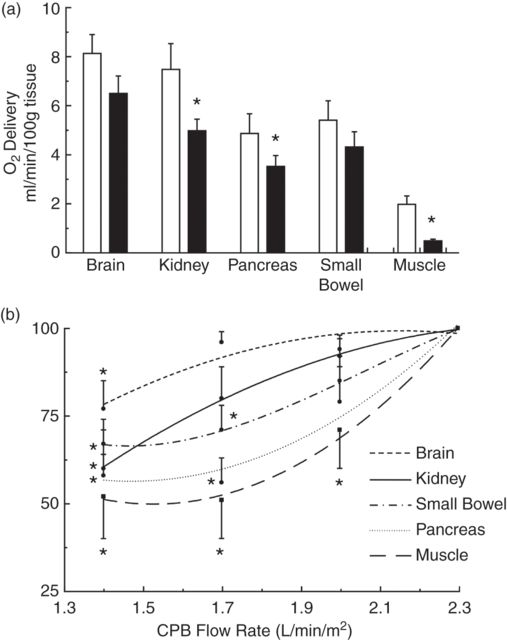
(a) Regional O2 delivery with onset of cardiopulmonary bypass. (Adapted from Boston and Cook. Ann Thorac Surg 2001.) (b) Change in organ DO2 with changes in CPB flow rate.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


