Chapter 82 Non-antiarrhythmic Therapies for Cardiac Arrhythmias
Background
Heart disease in any form may predispose the patient to the development of ventricular arrhythmias, some of which are lethal and may lead to sudden cardiac death (SCD). As a major public health burden, SCD has an incidence of nearly a half-million cases every year in the United States.1 Considerable effort has been invested in devising therapies specifically aimed at reducing the incidence of ventricular arrhythmias, particularly in the setting of ischemic heart disease and heart failure, where these fatal arrhythmias are exceedingly common. To date, however, the preponderance of evidence shows that the strategy of using antiarrhythmic drug therapy to directly suppress arrhythmias fails to improve morbidity or mortality outcomes. In ischemic heart disease, the Cardiac Arrhythmia Suppression Trial (CAST), the Canadian Amiodarone Myocardial Infarction Arrhythmia Trial (CAMIAT), and the European Myocardial Infarct Amiodarone Trial (EMIAT) found that antiarrhythmic therapy either increased mortality or failed to reduce it.2–4 A similar observation was noted in the Survival Trial of Antiarrhythmic Therapy in Congestive Heart Failure (CHF-STAT) and the Antiarrhythmic Trial with Dronedarone in Moderate to Severe Congestive Heart Failure Evaluating Morbidity Decrease (ANDROMEDA) in the setting of heart failure.5,6 The use of an implantable cardioverter-defibrillator (ICD), a form of nonpharmaceutical antiarrhythmic therapy, has been shown to be beneficial in the long term but has failed to improve survival in the early postmyocardial infarction period when the incidence of SCD is highest.7
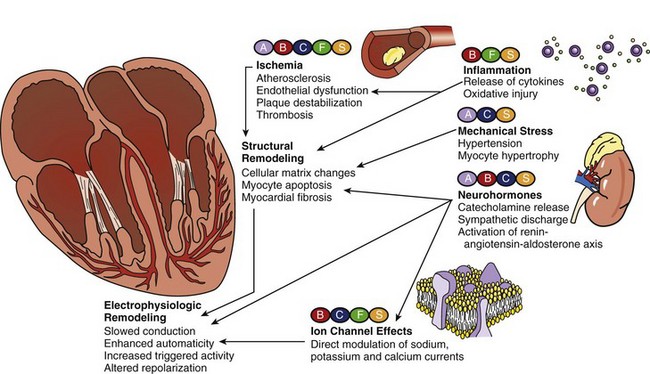
Growing evidence, however, suggests that certain therapies that are not used primarily for antiarrhythmic intent can actually be effective in preventing the development of ventricular arrhythmias, reducing their recurrence, and decreasing SCD incidence. Among these so-called non-antiarrhythmic therapies are β-adrenergic blocking agents, calcium channel blockers (CCBs), statins, fish oil, and renin-angiotensin-aldosterone system (RAAS) antagonists, and others (Figure 82-1). These agents are thought to target the underlying disease processes and reverse the basic pathophysiological substrates that promote arrhythmogenesis; this is expected to result in long-term cardiac electrophysiological stabilization.
Sudden Cardiac Death and Ventricular Arrhythmias
SCD is the unexpected natural death from a cardiac cause a short time after onset of symptoms, defined epidemiologically as any cardiac death occurring out of the hospital or taking place in the emergency room. It accounts for 63% of all cardiovascular deaths, and 84% of SCDs are caused by ventricular arrhythmias, often starting as ventricular tachycardia, rapidly progressing to ventricular fibrillation.8,9 Although not all instances of SCD are caused by arrhythmias, it is used by most investigations on cardiovascular outcomes as a surrogate marker for lethal ventricular arrhythmias. Crude and imprecise as it may be, SCD incidence still serves as an important indicator that helps measure the clinical effects of various antiarrhythmic therapies.
β-Blockers
The role of β-adrenergic blocking agents in the prevention of ventricular arrhythmia and SCD is well established, especially in the setting of ischemic heart disease and heart failure. Early observational studies showed that the β-blockers timolol, metoprolol, and propranolol reduced the incidence of SCD by 30% to 45% after myocardial infarction. In a large randomized study, the Clopidogrel and Metoprolol in Myocardial Infarction Trial: The Second Chinese Cardiovascular Study (COMMIT-CCS2) found a 17% relative risk reduction in the incidence of ventricular fibrillation with the early use of metoprolol in patients with myocardial infarction.10 The Carvedilol Post-Infarct Survival Control in Left Ventricular Dysfunction (CAPRICORN) trial involving patients with myocardial infarction and heart failure also revealed that carvedilol was associated with a 77% reduction in the risk of malignant ventricular arrhythmias as well as a 23% reduction in overall mortality. In patients with heart failure, β-blockers were found to have a more pronounced and more consistent effect in reducing SCD than any other therapy, including angiotensin-converting enzyme (ACE) inhibitors. The Cardiac Insufficiency Bisoprolol Study II (CIBIS II) demonstrated that bisoprolol reduced SCD by 44% and total mortality by 34% in patients with severe symptomatic systolic heart failure. Similarly, β-blockers reduced SCD in heart failure by 43% in the Metoprolol CR/XL Randomized Intervention Trial in Chronic Heart Failure (MERIT-HF), in which long-acting metoprolol was used, and by 36% in the Carvedilol Prospective Randomized Cumulative Survival (COPERNICUS) trial, in which carvedilol was used.11 Some agents may have greater antiarrhythmic properties than others, as seen in the Carvedilol Or Metoprolol European Trial (COMET), where carvedilol reduced SCD incidence more than did metoprolol in patients with myocardial infarction.12
In ischemic heart disease, β-blockers reduce myocardial oxygen demand, optimize diastolic filling, and improve coronary blood flow as a result of their negative chronotropic and inotropic effects. Areas of ischemic myocardium are particularly prone to arrhythmogenesis, and the suppression of ventricular arrhythmias by β-blockers may be partly attributed to their anti-ischemic properties. Myocardial infarction induces a state of heightened catecholamine discharge and autonomic dysfunction, both of which lower the threshold for ventricular fibrillation. Antagonism of this high cardiac sympathetic tone may also explain the protective effect of β-blockers against SCD. Antiproliferative and antiatherogenic mechanisms have also been proposed for some agents. Studies on animal models of myocardial ischemia further suggest that certain β-blockers may have sodium channel–blocking actions that could stabilize the myocyte membrane and suppress ventricular arrhythmias, similar to class I antiarrhythmic agents.13 β-Blockers may even induce electrophysiological changes in action potential by blocking the L-type calcium current, the transient outward potassium current, and the rectifier potassium current, resembling the class III antiarrhythmic actions of amiodarone. Experimental models also postulate that specific β-blockers such as carvedilol could exert significant antioxidant activity that protects the myocardium from reperfusion arrhythmias through adenosine-mediated processes.14
Statins
In patients with atherosclerotic heart disease enrolled in the Antiarrhythmics versus Implantable Defibrillators (AVID) trial, lipid-lowering therapy was associated with a 40% relative risk reduction in ventricular tachyarrhythmias and cardiac mortality.15 This significant benefit was expressed early in follow-up and is unlikely to be from the effects on cholesterol levels. In the Multicenter Automatic Defibrillator Implantation Trial (MADIT)-II involving patients with coronary artery disease and heart failure, statin use was associated with risk reductions of 28% for ventricular arrhythmias and 35% for SCD, with no significant effect on non-SCD.16 In the setting of nonischemic cardiomyopathy, an analysis of the Defibrillators in Non-ischemic Cardiomyopathy Treatment Evaluation (DEFINITE) trial showed that statin therapy was associated with an 86% reduction in arrhythmic sudden death, 22% reduction in ICD shocks from ventricular arrhythmias, and 78% reduction in mortality.17 A meta-analysis of randomized controlled trials on pravastatin, simvastatin, atorvastatin, and fluvastatin showed that statin treatment decreased the risk of SCD by 19%.18 The findings of these observational studies were further supported by small randomized trials in which patients with advanced heart failure treated with statins had significantly lower rates of SCD and all-cause mortality but not of pump failure.19
Statins have pleiotrophic effects that go beyond lipid lowering, including anti-inflammatory, anti-proliferative, anti-oxidative, and anti-thrombotic properties, which may as well explain their apparent antiarrhythmic effects. Myocardial ischemia is a potent trigger for lethal ventricular arrhythmias, and both atorvastatin and pravastatin have been shown to significantly reduce myocardial ischemia in the Study Assessing Goals in the Elderly (SAGE) and the Regression Growth Evaluation Statin Study (REGRESS).20,21 Proposed mechanisms include inhibition of luminal narrowing, restoration of endothelial function, plaque stabilization, anti-inflammatory effects, as well as modification of hemostatic processes that ultimately lead to improvement in myocardial perfusion and amelioration of ischemia-driven reperfusion injury. Ischemic heart disease is associated with cardiac electrophysiological remodeling leading to the formation of anatomic barriers, regions of slow conduction, and prolonged repolarization. In animal models of myocardial infarction and heart failure, successful inhibition of myocardial remodeling has been demonstrated with simvastatin and fluvastatin.22,23
Aside from their anti-ischemic properties, statins may also exert antiarrhythmic effects through various structural, neurohormonal, and electrophysiological mechanisms. The bilayer phospholipid structure of the membranes, as well as its functions, can be altered by changes in the dietary distribution of fatty acids.24 At least in theory, it may be possible for statins to exert direct antiarrhythmic effects by changing the lipid environment of the myocyte cell or organelle membranes, which results in altered channel protein function and ionic currents. Cardiomyocyte hypertrophy usually follows myocardial infarction and contributes to ventricular remodeling that, in turn, predisposes to ventricular arrhythmias. Pravastatin has been shown to decrease such hypertrophy in animal studies, probably by decreasing the secretion of endothelin-1. Dysfunctional intramyocardial calcium handling results in ventricular action potential abnormalities. By inhibiting intracellular calcium mobilization and transmembrane currents, statin therapy could stabilize ventricular repolarization and decrease the risk of arrhythmia.25 Overexposure of the myocardium to cathecholamines, in addition to worsening ischemia, also directly stimulates arrhythmogenesis. In experimental models, lipid-lowering therapy has been shown to normalize autonomic function and decrease sympathetic tone, as well as increase the cardiac response to parasympathetic stimulation.26,27 Diminished heart rate variability, an electrophysiological marker of endothelial dysfunction, is a known predictor of susceptibility to arrhythmias and SCD. Rosuvastatin and simvastatin have been shown in animal models to improve heart rate variability, likely through the modulation of nitric oxide synthase function.28 Increased QT variability, another marker of vulnerability to lethal arrhythmias, has been shown to improve with atorvastatin and fluvastatin therapy.29
Despite its well-established beneficial effect on cardiovascular mortality and evidence of reduction of SCD, the antiarrhythmic role of statins is still under debate because of conflicting reports. In the setting of myocardial ischemia, high-dose atorvastatin did not reduce the rates of resuscitated cardiac arrest in the Incremental Decrease in End Points Through Aggressive Lipid Lowering (IDEAL) and Myocardial Ischemia Reduction with Aggressive Cholesterol Lowering (MIRACL) trials.30,31 In patients with stable coronary artery disease, statin therapy similarly did not reduce the incidence of resuscitated cardiac arrest with the use of atorvastatin in the Treat to New Targets (TNT) trial and simvastatin in the Scandinavian Simvastatin Survival Study (4S).32,33 In fact, the Anglo-Scandinavian Cardiac Outcomes Trial (ASCOT) showed a trend toward a higher incidence of malignant arrhythmias with atorvastatin in high-risk hypertensive patients.34 The small number of patients who presented with life-threatening arrhythmias in these negative studies may explain disparities in outcomes, but large randomized trials that address the issue directly are still lacking. For now, statin use for prevention of ventricular arrhythmias seems only to benefit patients who had acute myocardial infarction, those who underwent revascularization procedures, and those who received ICDs.
Fish Oil
Epidemiologic data, including that of the U.S. Physicians Health Study, indicate that ingestion of very-long-chain n-3 polyunsaturated fatty acids (n-3 PUFA), found in fish or fish oil, is associated with reduced cardiovascular mortality.35 This was further reinforced by clinical investigations, including the Diet and Reinfarction Trial, which showed that consumption of fatty fish was associated with a 30% reduction in all-cause mortality.36 This was largely thought to result from the stabilization of the atherosclerotic plaque by n-3 PUFA through its lipid-lowering, anti-thrombotic, and anti-inflammatory properties. The GISSI-Prevenzione trial found a 45% reduction in SCD in patients with previous malignant ventricular arrhythmias who consumed 1 g of n-3 PUFA daily.37 In this trial, the beneficial effect of n-3 PUFA was thought to be unrelated to its effects on the lipid profile because the reduction in SCD appeared very early, and no effect was observed on the rate of myocardial infarction. This finding was reaffirmed in the Cardiovascular Health Study, which noted that frequent fish consumption was associated with a 58% reduction in arrhythmic death without changing the risk of myocardial infarction.38 This led to the theory that n-3 PUFA acids may have antiarrhythmic and antifibrillatory properties that are independent of their antiatherosclerotic and anti-thrombotic effects.
Such hypothesis is also supported by animal and experimental evidence. In an animal model of ischemia-induced SCD, infusion of n-3 PUFA emulsion prior to coronary artery ligation prevented ventricular fibrillation by 75%.39 Long-term feeding studies in primate models showed that n-3 PUFA supplementation and successful incorporation of the fatty acids into the myocardial membrane significantly reduced myocardial vulnerability to arrhythmic stimuli induced by ischemia.40 Dietary n-3 PUFA also reduced drug-induced repolarization abnormalities and abolished ventricular arrhythmias in a rabbit model.41 These cardioprotective effects have been attributed specifically to eicosapentaenoic and docosahexaenoic acids, which make up the majority of n-3 PUFA in fish-derived fats.
The consistent reduction of cardiac mortality and SCD with n-3 PUFA in observational human studies and experimental investigations has brought to light a major role of fish oil in cardiovascular therapy. However, randomized controlled trials have so far failed to demonstrate a clinically relevant antiarrhythmic effect. The Fatty Acid Antiarrhythmia Trial randomized patients at high risk for fatal ventricular arrhythmias to supplementation with either fish oil or placebo and found a trend toward reduced risk of arrhythmic events with n-3 PUFA, but this did not reach statistical significance.42 A multicenter, double-blind study in the United States that randomized patients with recent malignant ventricular arrhythmias to fish oil or placebo showed that n-3 PUFA supplementation did not prevent episodes of ventricular tachycardia or fibrillation.43 In fact, a trend toward increased ventricular arrhythmia occurrence was seen, which suggests a proarrhythmic effect of fish oil. The Study on Omega-3 Fatty Acids and Ventricular Arrhythmia (SOFA), a similar multicenter, randomized, placebo-controlled study conducted in Europe, also found no evidence of a strong protective effect of n-3 PUFA against ventricular arrhythmia.44 It did, however, establish that fish oil treatment does not cause harmful proarrhythmic effects. Meta-analysis of randomized trials involving nearly 33,000 patients found that fish oil supplementation was associated with a significant (20%) reduction in cardiac death, but it had no effect on arrhythmias or overall mortality.45 Proposed explanations for the inconsistent data in clinical trials include differences in study protocols, lack of standardized dosing, and inconsistencies in duration of supplementation, among a host of other confounders. Since most of the benefits were seen in the setting of early ischemic heart disease, it is conceivable that the antiarrhythmic utility of n-3 PUFA may be confined only to prevention of ventricular arrhythmia in patients with recent myocardial infarction, when the amount of scar tissue is still limited.
Renin-Angiotensin-Aldosterone System Antagonists
The benefit of ACE inhibitors in reducing cardiovascular and overall mortality in the setting of heart failure and ischemic heart disease is firmly established. A favorable effect on SCD was also noted, suggesting possible antiarrhythmic properties of ACE inhibitors. In patients with chronic heart failure, the Vasodilator Heart Failure Trial II (V-HEFT-I) demonstrated that enalapril reduced ventricular arrhythmias by 27% and SCD by 52%.46 Post hoc analyses of the Survival and Ventricular Enlargement (SAVE) trial and the Studies of Left Ventricular Dysfunction (SOLVD) trials also showed a trend toward reduced arrhythmic events and SCD, albeit not statistically significant.47,48 In patients with acute myocardial infarction, however, the Acute Infarction Ramipril Efficacy (AIRE) trial found that ramipril not only reduced total mortality but also decreased the incidence of SCD by 30%.49 Even in patients without heart failure, the Heart Outcomes Prevention Evaluation (HOPE) trial demonstrated that ramipril was able to cut arrhythmic death by 21%.50 Analyses of the Chinese Cardiac Study (CCS-1), the Cooperative New Scandinavian Enalapril Survival Study (CONSENSUS)-II, the Fourth International Study of Infarct Survival (ISIS-4), and the Survival of Myocardial Infarction Long-Term Evaluation (SMILE) trial also showed a trend toward reduced arrhythmic events and SCD, but none achieved statistical significance.51–54
Depressed left ventricle (LV) systolic function and ventricular dilatation are the most important predictors for developing ventricular arrhythmias. ACE inhibitors are thought to reduce SCD primarily because of their beneficial effect in reversing LV remodeling and improving systolic function. Electrophysiological remodeling also accompanies these myocardial fibrosis and structural changes, leading to slow conduction, early after-depolarization (EAD), and other proarrhythmic repolarization abnormalities. ACE inhibitors have been shown to reduce the degree of ventricular dispersion of repolarization by maintaining myocyte sodium and pH homeostasis, acting analogously as a class I antiarrhythmic agent.55 ACE inhibitors are also hypothesized to exert antiarrhythmic effects through antiatherosclerotic, anti-thrombotic, and neurohormonal mechanisms. Stabilization and regression of atherosclerotic plaque has been shown to be associated with ACE inhibitor therapy.56 Angiotensin-II is a vasoactive peptide that can activate inflammatory cytokines, promote ultrastructural myocardial fibrosis, modulate myocyte ion channels, induce release of endogenous cathecholamines, decrease gap junction conduction, and increase transmural dispersion of refractoriness in the ventricles.57 These effects substantially increase the propensity for arrhythmogenesis and SCD but can be effectively attenuated with ACE inhibitor or angiotensin receptor blocker (ARB) therapy.
Although observational and small prospective randomized studies point toward a modest reduction in ventricular arrhythmias, strong evidence to confirm the long-term antiarrhythmic benefit of ACE inhibitors is currently lacking.58 It was postulated that ACE inhibitors only have a modest effect on ventricular arrhythmias and SCD because of incomplete blockade of angiotensin II. In the Evaluation of Losartan in the Elderly (ELITE) study comparing the ARB losartan and the ACE inhibitor captopril in patients with heart failure, a 46% risk reduction in total mortality was seen with losartan compared with captopril, primarily because of a 64% risk reduction in SCD.59 This difference in SCD and mortality outcomes, however, was no longer seen in the subsequent ELITE-II trial. In the Optimal Trial in Myocardial Infarction with Angiotensin II Antagonist Losartan (OPTIMAAL) of patients with acute myocardial infarction, losartan was associated with a 19% risk reduction in SCD compared with captopril, although there were fewer cardiovascular deaths with captopril.60 ARBs were thought to exert a more complete inhibition of angiotensin II, especially at the tissue level, and this may explain its apparent advantage over ACE inhibitors. The Candesartan in Heart Failure Assessment of Reduction in Mortality and Morbidity (CHARM) trial showed that the ARB candesartan, when added to standard heart failure therapy that includes ACE inhibitors (with or without β-blockers and aldosterone-antagonists), improves mortality and morbidity outcomes, perhaps because of a more comprehensive inhibition of the RAAS.61 Like ACE inhibitors, regression of left ventricle hypertrophy (LVH) is seen with ARB therapy, which can explain a large part of its perceived antiarrhythmic effects. The Losartan Intervention For Endpoint Reduction in Hypertension (LIFE) trial found that regression of LVH with losartan was associated with a 30% reduction in SCD, independent of blood pressure reduction and other cardiac risk factors.62
Aldosterone can induce arrhythmogenesis through mechanisms similar to that of angiotensin-II, including detrimental effects on LV structural and electrophysiological remodeling, myocardial fibrosis, endothelial dysfunction, platelet activation, and sympathetic tone. In patients with chronic heart failure, the aldosterone antagonist spironolactone was associated with a significant reduction in SCD by 29% and total mortality by 30% in the Randomized Aldactone Evaluation Study (RALES).63 In patients with myocardial infarction, the Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS) found a 21% reduction in SCD and 15% reduction in overall mortality with the selective aldosterone blocker eplerenone.64 Eplerenone reduced SCD by 37% within 30 days when given early (7 days average) after myocardial infarction.65 Concerns have been expressed, however, regarding the safety of aldosterone antagonist therapy because of significant increase in hyperkalemia-related morbidity after publication of the RALES study.66
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


