CHAPTER 63 Myocardial Protection
Despite meticulous adherence to presently known principles of myocardial protection, perioperative myocardial damage related to ischemia-reperfusion injury continues to occur after cardiac operations that have been performed in a technically adequate manner. Ischemia-reperfusion injury associated with surgically induced myocardial ischemia secondary to aortic cross-clamping results from the attenuation or cessation of coronary blood flow such that oxygen delivery to the myocardium is insufficient to meet basal myocardial requirements to preserve cellular membrane stability and viability. Recovery after surgically induced ischemic arrest involves (1) resumption of normal oxidative metabolism and the restoration of myocardial energy reserves; (2) reversal of ischemia-induced cell swelling, loss of ion gradients, and adenine nucleotide losses; and (3) repair of damaged cell organelles such as mitochondria and the sarcoplasmic reticulum.
HISTORICAL DEVELOPMENT
After the initiation of open heart surgery with use of extracorporeal circulation by Gibbon,1 it soon became obvious that aortic cross-clamping was necessary to provide a bloodless field to facilitate the precise repair of intracardiac defects, to prevent air embolism when the left side of the heart was opened, and to avoid a turgid myocardium resistant to retraction. To overcome the difficulties of operating on a rheumatic mitral valve in a patient with aortic regurgitation, Melrose and colleagues2 introduced the concept of “elective cardiac arrest” by rapidly injecting into the aortic root, after aortic cross-clamping, a 2.5% potassium citrate solution in warm blood to arrest the heart. Soon thereafter, experimental and clinical evidence3 demonstrated the development of severe myocardial necrosis associated with the Melrose technique.
During the 1960s, two distinct technical pathways for management of ischemic arrest evolved. The “rapid operators” performing uncomplicated cases with short ischemic times adopted the use of normothermic ischemic arrest, until operative mortalities from the “stone heart” syndrome related to ischemic contracture of the myocardium4 associated with low levels of high-energy phosphate moieties became apparent.5 Intermittent aortic cross-clamping, involving reperfusion of the coronary circulation for 5 minutes after 15 minutes of ischemic arrest, is an empiric technique, still in use, and was based on the concept that after 15 minutes of ischemia, a sufficient concentration of high-energy phosphate moieties remained in the myocardium to allow replenishment of myocardial stores during the reperfusion period.6 Later studies demonstrated that there was no functional or metabolic advantage to intermittent reperfusion for normothermic ischemic periods up to 60 minutes.7,8 Nevertheless, intermittent cross-clamping accompanied by ventricular fibrillation continues to be used for coronary artery bypass surgery with results comparable to those of cardioplegic techniques.9
Fibrillatory Arrest
Electrically induced ventricular fibrillation with coronary perfusion was introduced by Glenn and Sewell10 and Senning11 as a means of avoiding air embolism. However, Buckberg and Hottenrott12 and Hottenrott and coworkers13 demonstrated subendocardial ischemia and necrosis with this technique, particularly in the hypertrophied ventricle. Later studies showed that if ventricular distention is avoided and coronary perfusion is maintained, postischemic fibrillation is not deleterious in the nonhypertrophied heart.14 Further validation of this technique, modified by mild hypothermia and the avoidance of aortic cross-clamping, has produced comparable clinical results for coronary revascularization.15,16
Continuous Coronary Perfusion
In an attempt to mimic the physiologic state, continuous coronary perfusion with a beating heart at normothermia or mild hypothermia at 32°C to prevent the onset of ventricular fibrillation became the preferred technique of myocardial preservation in the late 1960s and early 1970s, particularly after the report by McGoon and colleagues17 of 100 consecutive aortic valve replacements without a mortality. If aortic valve regurgitation was present or aortic root surgery was performed, the heart was kept beating by perfusing the individual coronary arteries with cannulas inserted into the ostia. However, in reality, continuous perfusion became intermittent as coronary perfusion was often discontinued to achieve better visualization of the operative field during critical portions of the procedure.18 In addition, problems with the coronary cannula, such as poor fixation, leaking associated with calcified ostia, early division of the left main coronary artery resulting in high perfusion pressure necrosis, and damage to the coronary artery such as dissection and late stenosis, continued to occur.19,20 Nevertheless, the technique of continuous coronary artery perfusion, either during the performance of complex aortic root surgery or for special situations such as re-do mitral valve surgery through a right thoracotomy incision after previous coronary artery revascularization with arterial conduits, continues to be used.21
Hypothermia
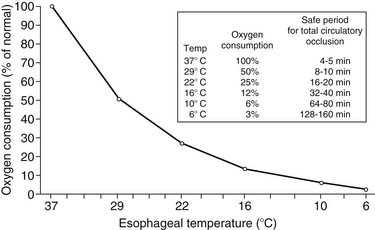
The earliest attempts to perform open heart surgery before the advent of the heart-lung machine used systemic hypothermia produced by surface cooling not only to protect the heart but to protect the brain and other organs during circulatory arrest.22,23 Hypothermia protects the ischemic myocardium by decreasing heart rate, slows the rate of high-energy phosphate degradation,24 and decreases myocardial oxygen consumption (Fig. 63-1).25,26 However, uniform cardiac hypothermia is difficult to achieve solely by the introduction of cold intracoronary perfusates, and systemic hypothermia is necessary, particularly in the presence of coronary obstruction, ventricular hypertrophy, rewarming of the right ventricle by the liver’s acting as a “heat sink,” and environmental rewarming.27 In an attempt to overcome this problem, Shumway and associates28 introduced the concept of profound local (topical) hypothermia by filling the pericardial sac with ice-cold saline.29 Although this technique is still used as an adjunct to other methods of myocardial protection, it is rarely used as the sole protective method because of the problem of warm bronchial collateral flow reaching the heart cavity, resulting in transmyocardial temperature gradients and resultant ischemia.30,31
Reintroduction of Cardioplegia
While cardioplegia had been abandoned for alternative techniques in the United States after the adverse experience with the Melrose potassium solution, Bretschneider,32 in Germany, continued studying induced cardiac arrest with use of a sodium-poor, calcium-free, procaine-containing solution (Bretschneider solution). Clinical application soon followed by Kirsch and asssociates33 using a magnesium aspartate–procaine solution. Hearse and coworkers34 introduced the concept of using an extracellular rather than an intracellular solution (St. Thomas’ solution), which was first applied clinically by Braimbridge and coworkers.18 On the basis of improved clinical outcomes, North American investigators35–38 initiated experimental studies using potassium cardioplegia followed by clinical reports39,40 demonstrating the efficacy of cardioplegia. During the next 3 decades, numerous investigators have continued their “quest for ideal myocardial protection.”41–44
BIOLOGY OF SURGICALLY INDUCED MYOCARDIAL ISCHEMIA
Global ischemia is not an all-or-nothing phenomenon; rather, it is heterogeneous in that at any moment of time, myocardial cells will have varying extents of damage.45,46 These changes affect cellular metabolism, ion transport, electrical activity, contractile function, vascular responsiveness, tissue ultrastructure, changes in nuclear and mitochondrial DNA, release of free radical oxygen species, and activation of inflammatory components.
Myocardial Oxygen Consumption
Because the heart is an obligate aerobic organ, it depends on a continuous supply of oxygen to maintain normal function. Myocardial oxygen reserve is exhausted within 8 seconds after the onset of normothermic global ischemia.47 Myocardial oxygen consumptionis compartmentalized into the oxygen needed for external work of contraction (80% to 90%) and the unloaded contraction, such as basal metabolism, excitation-contraction coupling, and heat production.48 A unique aspect of myocardial energetics is that 75% of the coronary arterial oxygen presented to the myocardium is extracted during a single passage through the heart; thus, depressed coronary venous oxygen content persists despite a wide range of cardiac workloads. Therefore, the heart is susceptible to the limitations of oxygen delivery, whereby an increase in 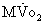 can be met only by augmentation of coronary blood flow. This is diametrically opposite to skeletal muscle, in which increased oxygen demand can initially be met by an increase in oxygen extraction. Clinically, a marked increase in coronary blood flow is observed at the beginning of the reperfusion period, after the aortic clamp is removed.
can be met only by augmentation of coronary blood flow. This is diametrically opposite to skeletal muscle, in which increased oxygen demand can initially be met by an increase in oxygen extraction. Clinically, a marked increase in coronary blood flow is observed at the beginning of the reperfusion period, after the aortic clamp is removed.
Biochemical Alterations
Under aerobic conditions, the heart derives its energy primarily from mitochondrial oxidative processes, using a variety of substrates such as glucose, free fatty acids, lactate, pyruvate, acetate, ketone bodies, and amino acids.49,50 However, oxidation of fatty acids provides the major source of energy production and is used in preference to carbohydrates. As tissue PO2 falls, oxidative phosphorylation, electron transport, and mitochondrial adenosine triphosphate (ATP) production cease. Early in ischemia, the heart depends on the energy production of glycogenolysis and aerobic glycolysis (Pasteur effect). Reduced mitochondrial activity leads to the accumulation of glycolytic intermediaries, reduced NADH, and the reduction of pyruvate to lactate. The resultant severe intracellular acidosis impairs contractile function, enzyme transport, and cell membrane integrity. This results in a cellular loss of potassium and pathologic accumulation of sodium, calcium, and water (Fig. 63-2).51
Ischemia-Reperfusion Injury
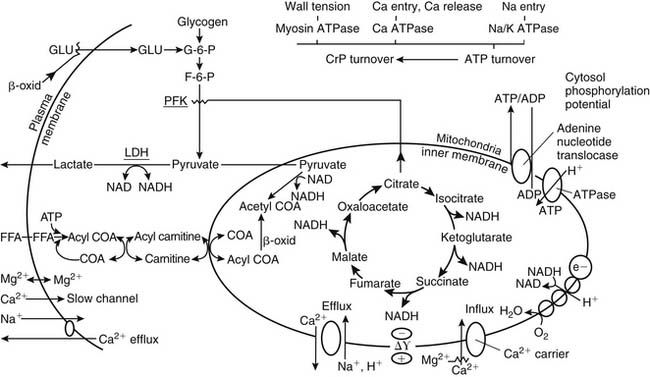
Ischemia-reperfusion injury occurs as the result of attenuation or cessation of coronary blood flow such that oxygen delivery to the myocardium is insufficient to meet basal myocardial oxygen requirements to preserve cellular membrane stability and viability. The initiation of myocardial injury requires an ischemic episode that, by itself, may induce reversible or irreversible cellular injury.52
Reversible ischemia-reperfusion injury may be manifested as either stunning or hibernation. Stunning “describes the mechanical dysfunction that persists after reperfusion despite the absence of myocellular damage and despite the return of normal or near-normal perfusion.”53,54 A second form of reversible ischemia-reperfusion injury is hibernation, which is a syndrome of reversible, chronically reduced contractile function as a result of one or more recurrent episodes of acute or persistent ischemia, referred to as chronic stunning.
As in stunning, hibernating myocardium is viable but not functional and is reversible with coronary revascularization.55,56 There is good clinical evidence that despite seemingly adequate application of modern methods of myocardial protection, all patients undergoing cardiac surgery have varying degrees of myocardial stunning.57,58 Evidence to support this concept is based on the requirement of inotropic support for separation from bypass for hours or days after surgery in some patients who are eventually weaned from these drugs as the stunning abates, without objective evidence of a myocardial infarction.59
Two major theories have been proposed as possible mechanisms leading to ischemia-reperfusion injury (Fig. 63-3).
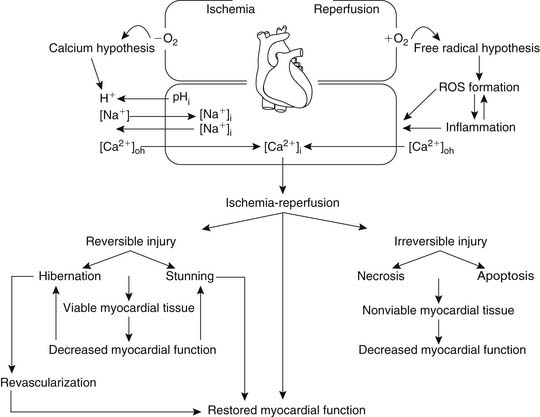
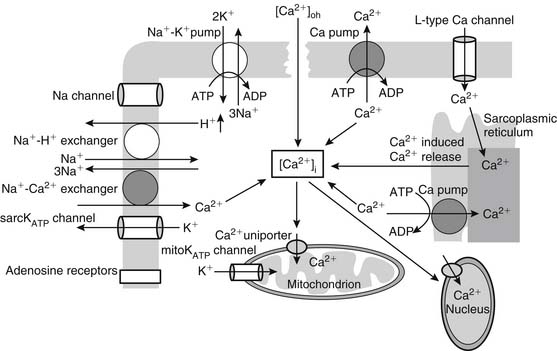
The calcium hypothesis suggests that the inability of the myocyte to modulate intracellular and intraorganellar calcium homeostasis induces a cascade of events culminating in cell injury and death (Fig. 63-4). Ischemia leads to the induction of metabolic acidosis and the activation of the sodium-proton exchanger, resulting in the transport of hydrogen ions to the extracellular space and the movement of sodium into the cytosol. As the sodium-calcium exchanger is activated, sodium is transported to the extracellular space and calcium is taken up into the cytosol, increasing cytosolic calcium concentration ([Ca2+]i). Increased [Ca2+]i accumulation is also augmented by ischemia-induced depolarization of the membrane potential, which allows the opening of the L-type calcium channels and further calcium entry into the myocyte. Cellular and cytosolic calcium-dependent phospholipases and proteases are in turn activated, inducing membrane injury and the further entry of calcium into the cell. These processes alter myocardial cellular homeostasis, leading to cellular dysfunction or, if they are of sufficient duration or intensity, cell injury or death. Alternative explanations include the concept of reperfusion-induced myocardial contracture resulting from rapid re-energization of contractile cells with persistent calcium overload affecting myofibrillar calcium sensitivity.60
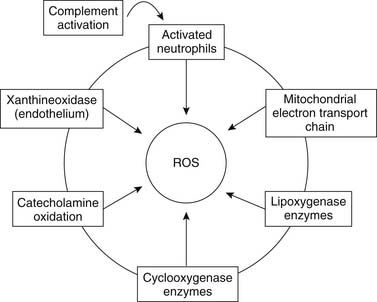
The free radical hypothesis suggests that the accumulation of partially reduced molecular oxygen collectively known as reactive oxygen species during the early stages of reperfusion causes myocardial cellular damage and cell death through microsomal peroxidation of the cellular phospholipid layer, leading to loss of cellular integrity and function (Fig. 63-5).61 The generation of reactive oxygen species is believed to be mediated by xanthine oxidase, activation of neutrophils, or dysfunction of the mitochondrial electron transport chain. It has been suggested that the generation of reactive oxygen species may induce cellular membrane damage, thus facilitating calcium entry and the induction of cellular death. This hypothesis unifies both prevailing theories and is currently considered to be valid. However, therapeutic attempts to control calcium and reactive oxygen species overload have not yielded meaningful advantage.
Alternative explanations include the concept of lethal reperfusion injury, defined as death of myocardial cells that were viable immediately before reperfusion.62 In an excellent review by Yellon and Hausenloy,63 alternative cardioprotective strategies are suggested to manage this injury by reperfusion injury salvage kinase pathways and targeting mitochondrial permeability transition pores to avoid mitochondrial calcium overload. Cyclosporine, a potent inhibitor of mitochondrial permeability transition pores, has recently been shown to limit infarct size after percutaneous coronary intervention during acute myocardial infarction.64
Irreversible Cell Injury
Irreversible cell injury, described ultrastructurally by Schaper and coworkers,46 occurs by two morphologically distinct pathways, necrosis and apoptosis. Necrosis is initiated by noncellular mechanisms with cell swelling, depletion of ATP stores, and disruption of the cellular membrane involving fluid and electrolyte alterations.65 In contrast, apoptosis or programmed cell death66 is an evolution-based mode of cell death characterized by a discrete set of biochemical and morphologic events involving the regulated action of catabolic enzymes (proteases and nucleases) that results in the ordered disassembly of the cell, distinct from cell death provoked by external injury.67
Inflammation
Inflammation has been implicated as a secondary mechanism contributing to injury after reperfusion. It is initiated through complement activation leading to the sequential formation of a membrane attack complex, which creates a cellular lesion and eventual cell lysis.68 In addition, cytokines, vasoactive and chemotactic agents, adhesive molecule expression, and leukocyte and platelet activation participate in the inflammatory process, producing cytotoxic molecules that facilitate cell death.69,70 Oxygen-derived free radical scavengers have also been used to limit reperfusion injury.71 Tissue factor, an inflammatory and procoagulant mediator, initiates the extrinsic coagulation cascade, resulting in thrombin generation and fibrin deposition, and may be related to the no-reflow phenomenon.72 Clinical applicability of anti-inflammatory agents awaits well-designed clinical trials because studies up until now have not shown any “meaningful cardioprotective effect.”63,73 Endothelium-dependent microvascular responses and coronary artery spasm may be related to reduced myocardial perfusion after reperfusion.74
Effects of Age
The vulnerability of the heart to ischemia-reperfusion injury is altered with temporal development. The newborn heart is more resistant to the effects of ischemia-reperfusion, which may be related to developmental differences in calcium transport and sequestration, and it is better able to restore myocardial function and myocardial high-energy phosphate stores after an ischemic event.75–77 In the adult heart, functional recovery is significantly delayed, and the recovery of high-energy phosphate stores is slower in returning to preischemic levels.78 As the heart ages, there are anatomic, mechanical, ultrastructural, and biochemical alterations that compromise the adaptive response of the heart.79 As a result, the senescent myocardium is less tolerant than the mature myocardium to surgically induced ischemia.80 The susceptibility of the aged myocardium to ischemia-induced injury is evident at many levels. Morphologically, with age, left ventricular mass is increased, as is a reduction in the size of the left ventricular cavity, accompanied by increased calcification of the valve anulus and coronary arteries.81 Ultrastructurally, there is decreased mitochondria to myofibril ratio, cardiac myocyte enlargement, and loss of mitochondrial organization as well as alteration in myocardial contractile properties.82 As a consequence of these changes, cardiac surgical operative mortality increases with age.83
Cyanosis
Cyanosis significantly increases the vulnerability of the myocardium to ischemia-reperfusion injury.84,85 However, cyanotic myocardial cells exhibit normal metabolism when they are provided with adequate oxygen and substrate.86 Mortality rates in patients undergoing elective repair for tetralogy of Fallot are related to the method of myocardial protection as well as to the patient’s age, duration and extent of cyanosis, and extent of hypertrophy.87
Ventricular Hypertrophy
Increased myocardial mass is an adaptive response to prolonged increases in myocardial workload due to pressure or volume overload. If it is untreated, progressive ventricular hypertrophy results in ventricular dilation and contractile dysfunction.88 Hypertrophied hearts have an increased vulnerability to ischemic injury, which has been attributed to accelerated loss of high-energy phosphate moieties,89 increased accumulation of lactate and hydrogen ions, earlier onset of ischemic contracture, and accelerated calcium overload after reperfusion.78,90,91 With ventricular hypertrophy, epicardial coronary arteries dilate in response to increased oxygen demands, while there is a decreased capillary density and vascular dilation reserve in the subendocardial regions resulting in increased ischemic vulnerability.92 Subendocardial ischemia leading to necrosis can occur during periods of hypotension, inadequate cardiopulmonary bypass, and ventricular fibrillation.93 The hypertrophied heart is particularly susceptible to ischemic injury in the early postoperative period, when hypotension associated with surgically induced myocardial stunning, hypothermia, and vasoconstrictor agents are present.
CARDIOPLEGIA: BASIC PRINCIPLES
A rational approach for protecting the heart during surgically induced ischemia not only must focus on the requirements for sustaining the ischemic cell but also must be compatible with the technical aspects of the operative procedure. Operative procedures require a flaccid arrested heart, a bloodless operative field, and sufficient time for the satisfactory repair of complex cardiac defects. Moreover, the ability of the heart to assume normal electromechanical function adequate to support the systemic circulation must rapidly follow the ischemic interval. The need for inotropic support or mechanical support devices (e.g., intra-aortic balloon assist device, ventricular assist device) to wean the patient from cardiopulmonary bypass when support was not required preoperatively represents a failure of myocardial protection. Studies demonstrate that dopamine treatment of postischemic myocardial stunning induces apoptosis.94 Nevertheless, despite meticulous adherence to known principles of protection, these events not infrequently and randomly occur and represent the limitations of present knowledge. Most investigators agree that the basic principles for adequate myocardial protection include (1) rapid induction of arrest, (2) mild or moderate hypothermia, (3) appropriate buffering of the cardioplegic solution, (4) avoidance of substrate depletion, and (5) attention to intracellular edema.95–97
Rapid Cardiac Arrest
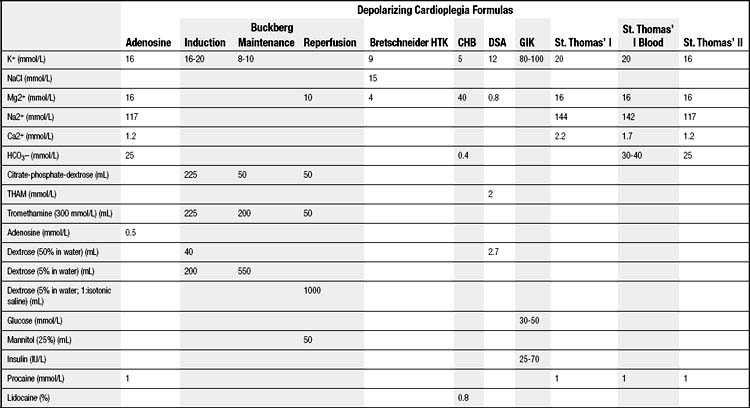
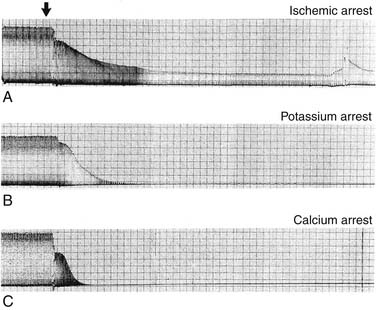
Rapid cardiac arrest remains the mainstay of adequate myocardial protection and is “achieved by targeting various points in the excitation-contraction coupling pathway” (Fig. 63-6).98 The induction of immediate cardiac arrest after the aorta has been clamped minimizes the depletion of high-energy phosphate moieties by useless mechanical work (Table 63-1). Potassium is the most common agent used for chemical cardioplegia and produces rapid diastolic arrest (Fig. 63-7). As the extracellular potassium concentration increases, the resting myocardial cell membrane becomes depolarized; the voltage-dependent fast sodium channel is inactivated, arresting the heart in diastole; and the slow calcium channel is activated, resulting in cytosolic calcium overload.99,100 The optimum concentration of potassium is thought to vary between 15 and 40 mmol/L,101 although it has been suggested that concentrations exceeding 20 mmol/L promote calcium overload and subsequent injury.102 Because the heart will remain arrested until the concentration of extracellular potassium or other cardioplegic ingredient is decreased by noncoronary collateral mediastinal blood flow, re-infusions of cardioplegia are necessary every 15 to 30 minutes.103 Clinical studies indicate that despite the infusion of large volumes of hyperkalemic cardioplegic solution for complex procedures requiring prolonged ischemic times, serum potassium levels in patients with normal renal function rarely exceed 5.5 mEq/L, as increased urinary excretion of potassium rapidly compensates for the endogenous administration of potassium.104
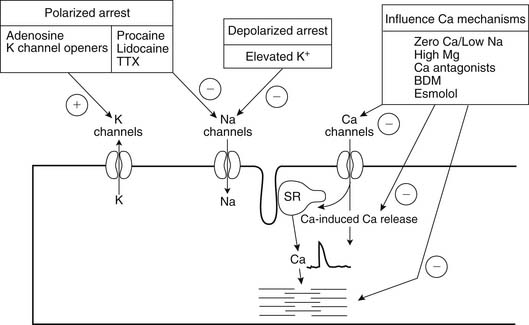
Agents inducing polarized arrest, in which the cell membrane potential remains close to resting potential, have significant advantages by limiting ionic movement and thereby reducing myocardial energy use.98 Sodium channel blockade, which arrests the heart by preventing the rapid sodium-induced depolarization of the action potential, includes procaine105 and tetrodotoxin.106 This class of drugs has been used successfully experimentally but is rarely used clinically at present. Potassium channel openers induce arrest by membrane hyperpolarization, couple the membrane potential to the cellular metabolic status, and afford cardioprotection by a similar mechanism associated with ischemic preconditioning.107 Two potassium–adenosine triphosphate (KATP) channel subtypes have been shown to coexist in the myocardium; one subtype is located in the sarcolemma (sarcKATP) membrane and the other in the inner membrane of the mitochondria (mitoKATP), and they can be pharmacologically manipulated by openers and blockers.96 Potassium channel openers have been used in conjunction with hyperkalemic and magnesium-containing cardioplegic solutions, although their clinical use remains controversial.108,109 However, mitochondria-specific potassium channel openers, such as diazoxide, have been demonstrated to have potential benefits when they are used with magnesium-supplemented potassium cardioplegia.110,111
Adenosine is an endogenous nucleoside formed as a consequence of the breakdown of high-energy phosphate and is rapidly phosphorylated by adenosine kinase to adenosine monophosphate and incorporated into the high-energy phosphorus pool or deaminated by adenosine deaminase, present in erythrocytes, to inosine, which is transported from the cell.112 Extracellular adenosine is cleared through cellular uptake, primarily by erythrocytes and vascular endothelial cells, and has a reported half-life of less than 10 seconds in whole blood, which limits its use during a prolonged period of surgically induced ischemia.113 Adenosine induces hyperpolarized cardiac arrest by antagonizing calcium channels and has been shown to inhibit both the sinoatrial and the atrioventricular nodes and atrial myocardial contraction.114 Adenosine, by its ability to antagonize the direct depressant effects on both the sinoatrial and atrioventricular nodes and atrial tissue, results in sinus slowing and arrest. Clinically, adenosine has been used as a pretreatment before the initiation of cardiopulmonary bypass, when it has been shown to increase postoperative cardiac index and to reduce creatine kinase release115; as an arresting agent by bolus infusion116; and as an additive to potassium cardioplegia, when it reduces the time to arrest117 and reduces potassium-induced cytosolic calcium overload.118 It may also improve functional recovery when it is infused during the reperfusion period.119 Compared with the acellular St. Thomas cardioplegic solution, the addition of adenosine appeared to enhance postoperative cardiac function in a series of patients undergoing coronary revascularization.120 Phase II studies suggest that adenosine may reduce postoperative complications,121 although the results are open to question.122 Nevertheless, additional clinical trials are suggested.123
Hypothermia
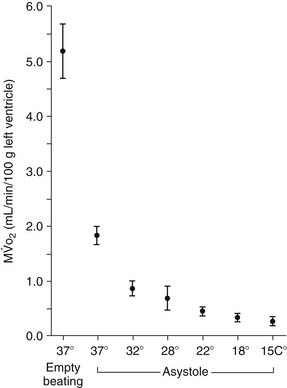
Hypothermia, whether it is mild (tepid at the room temperature range of 28°C to 32°C) or moderate (22°C to 25°C), continues to remain an indispensable adjunct for adequate myocardial protection. As noted in the Historical Development section, hypothermia decreases the rate of the metabolic degradation of energy stores during surgically induced ischemia. In addition, there is experimental evidence that there is a significant decrease of left ventricular myocardial oxygen consumption of the heart in the beating nonworking, fibrillating, and arrested states at 22°C compared with a myocardial temperature of 37°C (Fig. 63-8).124 However, there is minimal advantage in reducing the myocardial temperature below 22°C in that the myocardial oxygen consumption is decreased by only a minimal amount, from 0.31 mL at 22°C to 0.27 mL at 15°C per 100 g of left ventricular tissue per minute.125 Moreover, in the clinical setting, it is virtually impossible to maintain a uniform myocardial temperature below 22°C solely by the use of cold (4°C) intracoronary cardioplegia infusates accompanied by regional hypothermia, particularly in the presence of coronary obstructions, ventricular hypertrophy, and variations in mediastinal noncoronary collateral blood flow. In addition, the atrial and ventricular septa are warmed by systemic and pulmonary venous return, heat sinks such as the liver warm the base of the heart, and the anterior-situated right ventricle is warmed by the operative environment. Because of the lack of uniformity of myocardial temperatures in various myocardial segments, there is no correlation between myocardial tissue acidosis and temperature, leading to the recent abandonment by many surgeons of routine myocardial temperature monitoring during operative procedures.126
Buffering of the Cardioplegic Solution
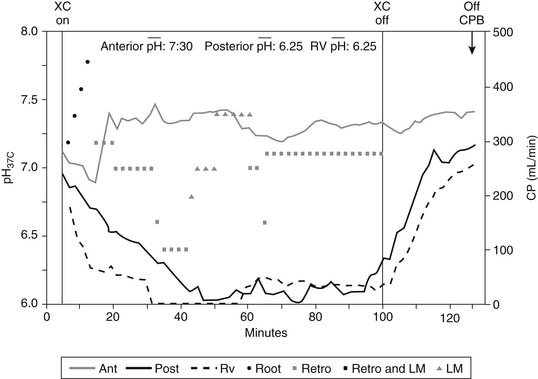
Buffering of the cardioplegic solution is necessary to combat the unremitting intracellular acidosis associated with surgically induced myocardial ischemia. Because the myocardium has the highest oxygen use of any organ in the body related to its concentration of mitochondria, ischemia results in the rapid accumulation of hydrogen ions and the reduction of intracellular pH, which has been quantified by the measurement of tissue PCO2 and hydrogen ion with phosphorus P 31 nuclear magnetic resonance spectroscopy in the laboratory and a pH probe in the operating room.78,127 With the recent development of a myocardial tissue pH probe, there is clinical evidence that maintenance of the tissue pH of 6.8 or greater is associated with adequate myocardial protection (Fig. 63-9).128 Thus, frequent infusions of cardioplegia, every 15 to 20 minutes, are necessary to prevent intracellular acidosis from reaching irreversible metabolic levels. In addition, hypothermia assists in the neutralization of acidosis because pH rises 0.0134 unit for each degree decrease in degree centigrade.129 However, the infusion of hypothermic cardioplegia does not restore pHi to prearrest levels but rather prevents further deterioration of the pH. When intermittent warm blood cardioplegia is used, lengthening of the ischemic period progressively increases intracellular metabolic acidosis and is probably associated with increased injury during repeated episodes of normothermic arrest.130,131 Bicarbonate, phosphate, aminosulfonic acid, tris-hydroxymethylamino-methane (THAM), and histidine buffers have all been used as cardioplegia additives to modulate pH.
Avoidance of Myocardial Edema
Avoidance of myocardial edema by controlling osmolarity is important to control volume regulation of the fluid compartments of the heart because myocardial edema is a known consequence of ischemia.132 The extent of myocardial edema has been shown to be directly modulated by osmolarity and onconicity of cardioplegia, with decreases being directly associated with increased myocardial edema and impaired diastolic filling.133 Hypotonic cardioplegic solutions cause myocardial edema134; hyperosmotic cardioplegia with an osmolarity in excess of 400 mOsm/L has been shown to cause myocardial dehydration.135 Isotonic solutions in the range of 290 to 330 mOsm/L or slightly hyperosmolar solutions appear to have the greatest clinical use, which is of particular importance in dealing with acellular cardioplegic solutions.40 Inert sugars including mannitol and sorbitol as well as metabolizable sugars such as glucose and dextrose have been used to increase osmolarity. However, when large volumes of continuous cardioplegia are administered, there is concern about producing hyperglycemia. Oncotic agents such as albumin and macromolecules, including dextrans and hydroxyethyl starches, have been used to prevent myocardial edema.136 Besides cardioplegic infusions, the hemodilution from crystalloid priming of the extracorporeal circuit, the activation of humoral and cellular mediators that increase microvascular permeability, and the impairment of myocardial lymphatic function may play major roles in the development of myocardial edema. Myocardial lymphatic function is dependent on the beating heart to transport fluid and is significantly reduced or completely stopped during cardiac arrest. Experimental evidence has indicated that during normothermic continuous antegrade blood cardioplegia, myocardial lymph flow is reduced to less than 20% of that in the normal beating heart.137
ALTERNATIVE ARRESTING AGENTS AND ADDITIVES
Beta-Blockers
As the ascending aorta is clamped, there is the nonexocytotic release of norepinephrine from the cardiac sympathetic nerves acting on β-adrenergic receptors on the outer surface of the sarcolemma, causing an increase in cAMP-dependent protein kinase activity, phosphorylation of the calcium release channels, and increased Ca2+ influx, resulting in increased Ca2+-dependent contractility and rapid depletion of glycogen stores.138 Early studies suggested that long-acting beta-blockers improved myocardial protection during ischemia but unfortunately have a prolonged negative inotropic effect, which limits their clinical use.139 For the most part, beta-blockers such as propranolol have been used as an adjunct to anesthesia to block β-adrenergic–stimulating episodes associated with coronary ischemia during the course of the operative procedure. Ultra-short-acting cardioselective beta-blockers such as landiolol and esmolol provide polarized cardiac arrest by maintaining the membrane potential at or near the resting membrane potential.140 The cardioselectivity of these agents is 50 to 250 times that of propranolol, and the half-life of esmolol is only 9 minutes because it is rapidly hydrolyzed by red blood cell esterase.141 Esmolol has been used to enhance myocardial protection during intermittent arrest142 and has been shown to provide myocardial preservation equivalent to or better than cold crystalloid or blood cardioplegia.143–145 With esmolol cardioplegia, there is a slow undulating ventricular contraction that may decrease myocardial edema but does not provide a quiescent operating field.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


