Chapter 43 Molecular Imaging Approaches for Evaluation of Myocardial Pathophysiology
Angiogenesis, Ventricular Remodeling, Inflammation, and Cell Death
INTRODUCTION
The application of imaging using biologically targeted markers (i.e., molecular imaging) has a number of requirements.1 A molecular target that adequately represents the process being studied is critical to the specificity of any imaging approach. The target must then lend itself to a readily synthesizable probe(s) that will bind to the target molecule with a high degree of specificity. Lastly, an imaging technology that provides the best combination of sensitivity and resolution (both spatial and temporal) to identify and localize the probe within the target organ system needs to be immediately available and economically feasible. Molecular imaging approaches are currently being developed for most of the imaging modalities, including nuclear, magnetic resonance, x-ray computed tomography (CT), optical fluorescence, bioluminescence, and ultrasound.2 Though each modality carries strengths and weaknesses, it is likely that the practical limitations of cost and widespread availability will determine which modalities become adapted for clinical use.
Single-photon emission computed tomography (SPECT) and positron emission tomography (PET) are imaging techniques that make use of radiolabeled probes and have been used for over 3 decades. Radiolabeling has the unique advantage of augmenting low signal-intensity objects. For example, PET can detect picomolar and nanomolar concentrations of a molecule of interest.2 Though SPECT offers the advantage of decreased cost and widespread availability, PET offers the advantages of increased sensitivity in conjunction with the ability to quantitate as well as repetitively image using tracers with ultrashort half-lives. In the past, nuclear imaging modalities have been limited by attenuation artifacts from soft tissue and partial volume effects. More recent systems combining CT imaging with either SPECT or PET have allowed for attenuation correction, leading to improved imaging quantification and registration.
ANGIOGENESIS
Atherosclerosis is a chronic inflammatory disease that leads to the progression of lipid-laden vascular plaques.3,4 Monocyte adhesion and transendothelial migration result in activation and differentiation, with amplifying production of cytokines/chemokines, as well as foam cell development when these subendothelial macrophages endocytose oxidized lipid. The progression of atherosclerotic disease can lead to the development of chronic ischemia in areas of myocardium, secondary to poor perfusion through narrowed blood vessels. Subsequent signals in response to the ischemia may stimulate angiogenic and arteriogenic responses to restore perfusion.
Angiogenesis is defined as the process of sprouting new capillaries from preexisting microvessels.5 There is a great deal of interest in understanding the processes of angiogenesis in order to design therapeutic treatments that allow revascularization through an iatrogenic-stimulated angiogenic response. This angiogenic process often occurs in association with arteriogenesis, which represents a remodeling of larger preexisting vascular channels or collateral vessels feeding the new microvascular network. The goal for any myocardial revascularization strategy would be to initiate angiogenesis and arteriogenesis in manner that improves tissue perfusion. There is a large body of literature devoted to understanding these phenomena.6–8
Angiogenesis is stimulated by external processes such as ischemia, hypoxia, inflammation, and shear stress. Endothelial cells, smooth muscle cells, blood-derived macrophages, and circulating stem cells all play distinctive roles in the angiogenic process. There is the careful interaction of these cells with each other as well as within the tissue of extracellular matrix proteins. The process itself consists of a series of endothelial cell responses to angiogenic stimulation, such as degradation of extracellular matrix (ECM), budding from parent vessels, proliferation, migration, tube formation, and ultimately maturation and maintenance of the new vessel.9 Figure 43-1 outlines a schematic representation of the process of angiogenesis on an endothelial cellular level.
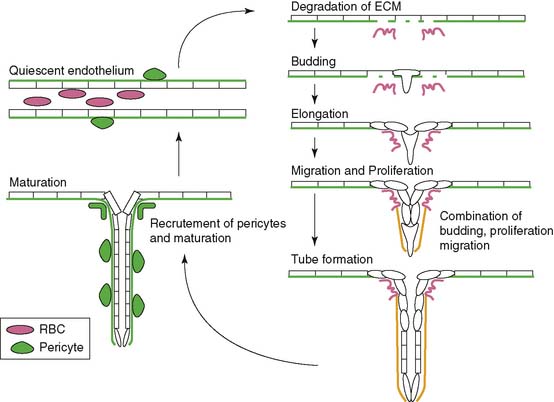
Hypoxia, the imbalance between oxygen delivery and demand in a given tissue, is a potent stimulator of angiogenesis.10 Hypoxic conditions such as myocardial ischemia from atherosclerotic disease or acute myocardial infarction (MI) result in up-regulation of the transcriptional activator hypoxia-inducible factor 1 (HIF-1).11 HIF-1 is a heterodimeric protein composed of alpha and beta subunits. It is continuously expressed and degraded through oxygen-dependent proline hydroxylation of the alpha subunit and subsequent ubiquitination by von Hippel-Lindau E3 ligase for degradation in the proteosome.12 Hypoxia stabilizes the HIF-1 protein by preventing the proline hydroxylation and ultimately the ubiquitination and degradation in the proteosome. This up-regulation of HIF-1 protein leads to the transcription of a number of hypoxia-inducible genes, including the key angiogenic mediators vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF) and TGF-β, and the VEGF receptors, Flt-1 (VEGFR-1) and FLK-1 (VEGFR-2).10,13–16
When VEGF binds to its receptors on the surface of endothelial cells, a signal is transduced through their tyrosine kinase activity. This initiates a series of processes that results in endothelial cell proliferation, migration, survival, and angiogenesis.17 Early work in this field generated enthusiasm for the prospect of therapeutic angiogenesis utilizing angiogenic proteins like VEGF or fibroblast growth factor 2 (FGF-2). In fact, a number of trials had been designed to attempt therapeutic angiogenesis, with the goals of stimulating new blood vessel growth and improving myocardial perfusion in ischemic heart disease. With relatively invasive measures to assess efficacy of treatment, preclinical studies in animal models had shown some benefit to therapeutic angiogenesis, supporting the transition to human trials.18 Based on the early results, clinical trials were designed and initiated.
The FGF-2 Initiating Revascularization Support Trial (FIRST) randomized patients with chronic angina (class III-IV) to three doses of bFGF protein intracoronary injection versus placebo.19 The study demonstrated no significant differences between the groups in exercise time, nuclear perfusion, or quality of life at 90 days. The Angiogenic Gene Therapy Trial (AGENT) randomized patients to a single escalating intracoronary dose of replication-defective adenovirus containing the FGF4 gene.20 There was no significant difference in exercise treadmill time at 4 and 12 weeks.
In contrast, the VEGF in Ischemia for Vascular Angiogenesis (VIVA) Trial randomized patients with inducible ischemia on myocardial perfusion imaging to receive two (low- or high-dose) intracoronary injections of VEGF-1 protein versus placebo, followed by three more injections on days 3, 6, and 9.21 There were no differences in exercise time or anginal class between the groups at 60 days, but anginal class was improved at 120 days in the high-dose group. There were no differences in myocardial perfusion throughout all the groups. The Randomized Evaluation of VEGF for Angiogenesis in Severe Coronary Disease (REVASC) Trial involved intramyocardial injection of replication-defective adenovirus containing the VEGF121 gene.22 This study demonstrated a clinical improvement in the gene-therapy patients that was sustained from 3 to 6 months. Patients had significant improvements in exercise treadmill time to an additional 1 mm of ST depression, exercise time to angina, and angina class; however, nuclear perfusion imaging supported the control group, with the caveat that the treated group achieve higher workloads overall. More recently, the Euroinject One phase 2 clinical trial involved intramyocardial injections of plasmid containing VEGF in patients with Canadian Class 3 and 4 angina.23 The study revealed no differences in perfusion abnormalities, but there were improvements in regional ventricular wall-motion disturbances and functional anginal class.
One important signaling molecule is tie-2, a receptor tyrosine kinase, primarily expressed on vascular endothelium. Tie-2 has two major ligands: angiopoietin-1, which plays an agonistic signaling role, and angiopoietin-2, which appears to play an antagonistic signaling role.24,25 In conjunction with VEGF, this signaling mechanism appears to help stabilize and mature new capillaries as they develop, providing an important dimension to endothelial signaling and effective therapeutic angiogenesis. Preliminary, animal studies support utilizing this multipronged approach to VEGF angiogenesis therapy to obtain a more functional vasculature.26
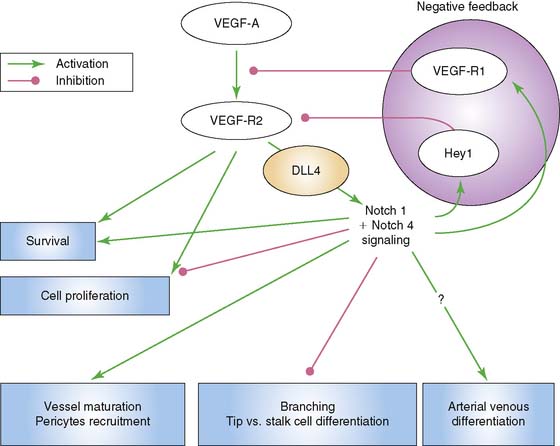
Another significant signaling system is that of the Notch–Delta-like ligand pathway. The Notch receptor is a single-pass transmembrane protein consisting of an extracellular domain that is responsible for ligand interaction, a transmembrane domain that is involved in receptor activation, and an intracellular signaling domain.9 Signaling through Notch requires cell to cell contact through binding of its ligand, which is also a cell-surface protein. The ligands are members of the Jagged and Delta-like ligand family. Of interest to vascular endothelial cells are Notch 1 and Notch 4, as well as their ligands, Jagged1, Delta-like ligand 1 (Dll1), and Delta-like ligand 4 (Dll4). Dll4 is found exclusively on endothelial cells. Murine gene disruption experiments have revealed that Notch-Dll4 signaling is crucial to normal embryonic vascular development.27 Blocking the Notch-Dll4 interaction can paradoxically lead to an increase in angiogenesis and new vessel budding, but the new vessels appear to function in an abnormal manner, which compromises blood flow and oxygenation to the area of interest.28–30 It appears that VEGF up-regulates Dll4 in endothelial cells in a manner that creates a complex feedback loop to help achieve functional new blood vessels (Fig. 43-2). This argues for the importance of understanding what molecular switches are being activated in any attempt at therapeutic angiogenesis.
Endothelial signaling through VEGF and its concomitant receptor pathways is not the only molecular event undertaken during angiogenesis. There is also up-regulation and activation of molecules like integrins that aid in the cell-cell signaling and ultimately extracellular remodeling process. Integrins are a family of heterodimeric (αβ) cell-surface receptors that mediate divalent, cation-dependent, cell-cell, and cell-matrix adhesion and signaling through tightly regulated interactions with their respective ligands.31 During angiogenesis, endothelial cells make use of integrins to adhere to one another and the extracellular matrix to construct and extend new vessels. Peak expression of one integrin in particular, αvβ3 integrin, has been shown to occur 12 to 24 hours after initiation of angiogenesis with FGF2.32 Integrins are capable of mediating an array of cellular processes, including cell adhesion, migration, proliferation, differentiation, and survival via a number of signal transduction pathways (Fig. 43-3).33,34 Activation of c-Jun NH2-terminal kinase (JNK) and extracellular signal-regulated kinase (ERK) may lead to endothelial cell–induced remodeling of the ECM in response to mechanical stimuli. Specifically, the endothelial cell integrin αvβ3 allows cells to interact with the ECM in a way that aids in endothelial cell migration.35 Through outside-in signaling, integrin αvβ3 also plays a critical roll in the survival of cells undergoing angiogenesis.32
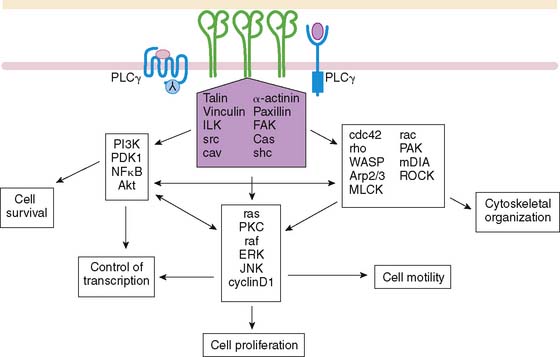
Several other molecules have been identified in this schema of endothelial cell activation. Syndecan-4 is a transmembrane heparan sulfate–carrying core protein that promotes binding of VEGF to VEGFR, resulting in activation of protein kinase C and downstream signaling.36 CD13 is a cell-surface antigen that is expressed in endothelial cells as an aminopeptidase—in other words, a membrane-bound metalloproteinase that appears to be essential for capillary tube formation.37 Degradation of the extracellular matrix involves up-regulation of matrix metalloproteinases, as well, to allow budding and expansion of the new vessel. Other molecular events include platelet-derived growth factor receptor signaling in response to HIF-1. This activates cell types like the perivascular pericytes or vascular smooth muscle cells, playing a crucial role in the new blood vessel development.38
RADIOTRACER-BASED IMAGING OF ANGIOGENESIS
Potential targets for molecular imaging of angiogenesis have been traditionally divided into three major categories: non-endothelial targets like molecules associated with monocytes, macrophages, and stem cells; endothelial cell targets like vascular endothelial growth factor (VEGF), integrins, CD13, and syndecan-4; and extracellular matrix proteins.39 Despite the wide array of molecules available as potential imaging targets for angiogenesis in response to ischemia, there are just two real active areas of research: imaging VEGF receptors via labeled VEGF and imaging the αvβ3 integrin via ligand-like analogs.
Vascular Endothelial Growth Factor Receptors
VEGF receptors have been targeted for imaging techniques in models of ischemia-induced angiogenesis. Radiolabeled-VEGF121 has been used to effectively identify angiogenesis in a rabbit model of hindlimb ischemia.40 In this study, KDR and Flt-1 receptor expression was increased in the immunohistochemistry analysis of the skeletal muscle, supporting the theoretical hypoxic-driven angiogenic response. Biodistribution of the radiotracer raises concern for the practical application of this system in humans. The biodistribution was 20-fold higher levels in critical organs (liver, kidneys) compared with ischemic limb and is presumably related to relative VEGFR density in these organ systems.
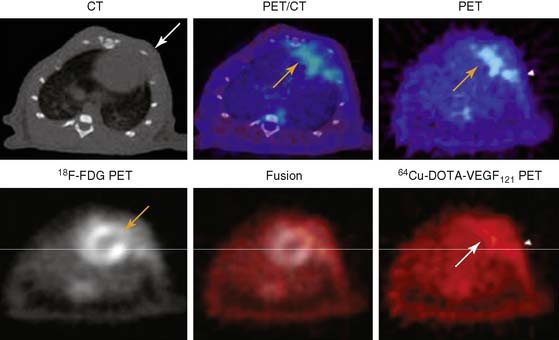
A PET tracer, copper-64-labeled (64Cu)-6DOTA-VEGF121, was recently developed for imaging angiogenesis in a rat model of MI (Fig. 43-4).41 Rats underwent ligation of the left coronary artery and subsequent PET imaging at various time points after MI. The investigators hypothesized that this tracer would detect early angiogenic signals because ischemia drives VEGFR expression. Co-registration of images was carried out using CT, and the zone of infarct was demonstrated using fluorine-18-labeled fluorodeoxyglucose (18F-FDG) uptake. The study demonstrated that 64Cu-6DOTA-VEGF121– specific signal was present in the infarct region and peaked on day 3, consistent with the changing levels of VEGFR expression in the tissue as analyzed by immunofluorescence microscopy. The same group has also published experiments supporting the use of 64Cu-6DOTA-VEGF121 for imaging of VEGFR-2 in a murine model of hindlimb ischemia-induced angiogenesis.42
Another type of cardiac-specific reporter has been developed as a gene expression system for use in rats, with microPET imaging (see Chapter 45 for specific details).43 Briefly, the system involves adenovirus delivery of mutated thymidine kinase under the control of a cytomegalovirus promoter driving expression in myocardial cells. The reporter probe is 18F-labeled fluoro-3-hydroxymethyl-butylguanine (18F-FHBG), which crosses the myocardial membrane and gets phosphorylated by the thymidine kinase. Phosphorylation essentially traps the 18F-FHGB in the myocardium for subsequent microPET imaging. Early studies revealed that the localized site of the mutated thymidine kinase, HSV1-sr39tk, corresponded closely with that defined by postmortem autoradiography, histology, and immunohistochemistry.44 Other studies have demonstrated the feasibility of utilizing a similar reporter system in pigs using a clinical PET scanner.45 The reporter system was then linked to VEGF to assess feasibility of developing an approach that links therapy and imaging.46 Early experiments with rat embryonic cardiomyocytes revealed a strong correlation in that both the mutated thymidine kinase and VEGF were expressed in the same cells. Further studies involved injection of the VEGF/thymidine kinase reporter system in models of ischemia. Using microPET, cardiac transgene expression was assessed and the in vivo imaging correlated well with ex vivo tissue studies for gamma counting, thymidine kinase activity, and VEGF levels. There appeared to be increased capillaries and small blood vessels in the VEGF-treated myocardium; however, there was no improvement in perfusion assessed by nitrogen-13 (13N)-ammonia imaging or metabolism assessed with 18F-FDG imaging. These studies suggest that a reporter system can be developed to help visualize the effectiveness of delivering VEGF gene therapies for stimulation of angiogenesis.
Integrin αvβ3
Imaging angiogenic vessels through targeting of αvβ3 integrin was first proposed through a series of magnetic resonance imaging studies using a monoclonal antibody to αvβ3 integrin tagged with a paramagnetic contrast agent.47 The studies were complicated by poor clearance of the tracer from the blood pool. Later studies made use of a number of αvβ3 antagonists that were radiolabeled.48,49 The experiments took advantage of the arginine-glycine-aspartate (RGD) binding sequence on integrins by synthesizing various RGD analogs.
An indium-111 (111In)-labeled quinolone (111In-RP748) revealed high affinity and selectivity for αvβ3 integrin in adhesion assays.50 Subsequent studies using a cy3-labeled homolog of 111In-RP748 demonstrated preferential binding to activated αvβ3 integrins on endothelial cells in culture, with localization to cell-cell contact points.51 Initial studies with this agent focused on imaging tumor angiogenesis, although the first imaging of ischemia-induced myocardial angiogenesis using 111In-RP748 was carried out in rat and canine models of infarction.52 In these studies, 111In-RP748 demonstrated favorable kinetics for in vivo SPECT imaging of ischemia-induced angiogenesis of the heart. Relative 111In-RP748 activity was markedly increased in the infarcted region acutely and persisted for at least 3 weeks post reperfusion.52,53 Therefore, targeted imaging with 111In-RP748 has demonstrated integrin αvβ3 activation early post infarction, suggesting a role for this technique in early detection of angiogenesis as well as for detection of chronic ischemia (Fig. 43-5).53
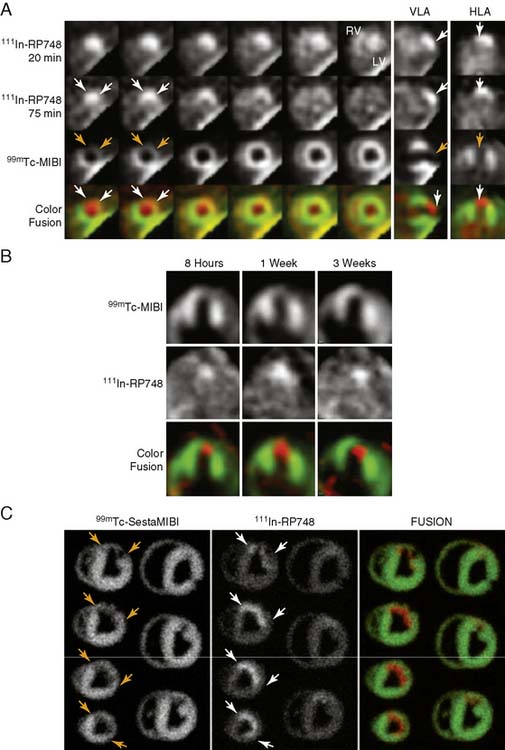
Figure 43-5 In vivo and ex vivo 111In-RP748 and 99mTc-sestamibi (99mTc-MIBI) images from dogs with chronic infarction. (A) Serial in vivo 111In-RP748 SPECT short-axis, vertical long-axis (VLA), and horizontal long-axis (HLA) images in a dog 3 weeks after LAD infarction at 20 minutes and 75 minutes after injection in standard format (Fig. 43-5A). 111In-RP748 SPECT, images were registered with 99mTc-MIBI perfusion images (third row). The 75-minute 111In-RP748 SPECT images were colored red and fused with MIBI images (green) to better demonstrate localization of 111In-RP748 activity within the heart (color fusion, bottom row). Right ventricular (RV) and left ventricular (LV) blood pool activity are seen at 20 minutes. White arrows indicate region of increased 111In-RP748 uptake in anterior wall. This corresponds to the anteroapical 99mTc-MIBI perfusion defect (orange arrow). (B) Sequential 99mTc-MIBI (top row) and 111In-RP748 in vivo SPECT HLA images at 90 minutes after injection (middle row) from a dog at 8 hours (Acute), and 1 and 3 weeks after LAD infarction (Fig. 43-5B). Increased myocardial 111In-RP748 uptake is seen in anteroapical wall at all three time points, although appears to be maximal at 1 week post infarction. Color fusion 99mTc-MIBI (green) and 111In-RP748 (red) images (bottom row) demonstrate 111In-RP748 uptake within 99mTc-MIBI perfusion defect. Ex vivo 99mTc-sestamibi (left) and 111In-RP748 (center) images of myocardial slices from a dog 3 weeks after LAD occlusion, with color fusion image on right (Fig. 43-5C). (C) Short-axis slices are oriented with anterior wall on top, RV on left. Orange arrows indicate anterior location of nontransmural perfusion defect region, and white arrows indicate corresponding area of increased 111In-RP748 uptake.
(From Meoli DF, Sadeghi MM, Krassilnikova, S et al: Noninvasive imaging of myocardial angiogenesis following experimental myocardial infarction, J Clin Invest 113:1684–1691, 2004. Reprinted with permission.)
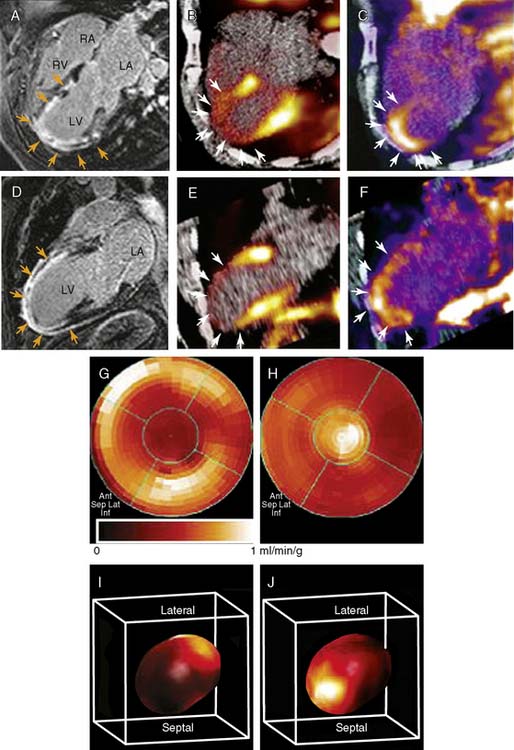
Other experiments, utilizing a technetium-99m (99mTc)-labeled peptide, NC100692, in the rodent model of hindlimb ischemia for targeting of αvβ3 integrin have also been carried out and support the value of integrin imaging in models of peripheral arterial disease.54 The recent imaging of αvβ3 integrin by the PET imaging tracer 18F-galakto-RGD in a 35-year-old patient with a transmural MI 2 weeks prior demonstrates the feasibility of detecting angiogenesis in the myocardium in humans (Fig. 43-6).55
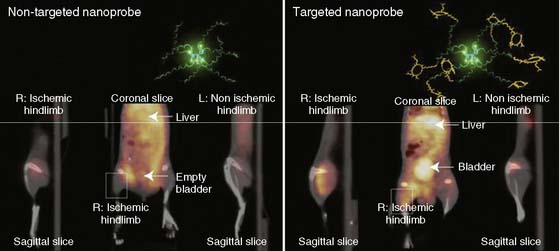
Recently, a new biodegradable positron-emitting nanoprobe targeted at αvβ3 integrin has been designed for noninvasive imaging of angiogenesis with a PET-based system (Fig. 43-7).56 The nanoprobe has a core-shell architecture that allows radiolabeling with radiohalogens that are linked to the core to protect them from dehalogenation. The terminal ends of the outer shell are covalently linked to cyclic-RGD peptides to confer specificity to αvβ3 integrin. This nanoprobe revealed enhanced binding of αvβ3 integrin using in vitro and cellular assays. The nanoprobe demonstrated favorable biodistribution when compared with untargeted probe in rodents, with a slight increase in uptake in phagocytic-rich organs like the spleen, liver, and kidneys. Using a bromine-76 (76Br)-labeled derivative of the nanoprobe, the investigators targeted αvβ3 integrin in a murine model of hindlimb ischemia. In vivo PET imaging revealed specificity of the probe to the ischemic limb, using the nonischemic limb and a version of the probe lacking the cyclic RGD peptides for controls. Ex vivo imaging and histologic analysis allowed for quantification of the radioactivity, leading to further association of the probe to areas of increased αvβ3 integrin expression and new vessel formation. The application of nanotechnology to angiogenesis imaging is an exciting new area of research, but further studies will be required to assess the feasibility of applying this system to the in vivo imaging of angiogenesis in the myocardium.
VENTRICULAR REMODELING
Ventricular remodeling is a complex biological process that involves inflammation, angiogenesis, repair, and healing, with specific biochemical and structural alterations in the myocardial infarct and periinfarct regions as well as remote regions (Fig. 43-8).57,58
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


