Introduction
The sequencing of the human genome is likely to be a landmark study of millennium proportions. The implications for cardiology from knowing the sequence of the human genome are many, but probably the most obvious is identifying the genes responsible for familial disorders. Abnormalities of the heart and blood vessels are number one among human birth defects occurring at about 1% of live births.1,2 Genetic diagnosis and management are expected to be incorporated into the practice of a cardiologist by the end of this decade.3 Knowing the etiology and understanding the pathogenesis of genetic disorders are most likely to improve the diagnosis, prevention and treatment of those disorders and in addition, provide fundamental insights into acquired disorders that simulate the phenotype. A good example is that of familial hypercholesterolemia in which there is a defective receptor for cell uptake of cholesterol.4 The findings confirmed that the plasma cholesterol was a major factor in coronary artery disease and subsequently led to unraveling of the synthesis, transport and degradation of cholesterol.
The standard treatment today for coronary artery disease, both familial and acquired, includes the use of statins to lower plasma levels of low-density lipoprotein (LDL) cholesterol. It must be emphasized that practically all genetic disorders have an environmental component and the resulting phenotype is due to an interplay between the gene (genotype) and the environment (phenotype). Furthermore, for single gene disorders it is now recognized that some of the variability in the phenotype is also due to interplay between the causal mutations, environmental factors and the modifier genes.5 An obvious example of the importance of environmental factors is that of familial hypertrophic cardiomyopathy (FHCM). FHCM is a single gene disorder that is transmitted in an autosomal dominant pattern, giving rise to a phenotype of left ventricular hypertrophy.6 The same genetic defect is present in the same abundance in the right and left ventricles yet the disease is seldom manifested in the right ventricle. This would imply that the high pressure of the left ventricle is an important stimulus in the pathogenesis of the phenotype of hypertrophy.
Genetic disorders are considered in three categories: chromosomal abnormalities, single gene disorders, and polygenic disorders. Chromosomal abnormalities are usually detected by the pediatric cardiologist in children while they are still very young. An example of an adult form of chromosomal abnormality would be Turner syndrome. In the last edition, we stated that the multigene disorders (e.g. coronary artery disease) are where the future must be but the technology had not yet arrived. The technology has now arrived and remarkable progress has been made in the past year.7–9
Mutations responsible for single gene disorders
Diseases due to an abnormality in a single gene are inherited in a predictable pattern termed Mendelian transmission. In single gene disorders, the responsible gene is both necessary and sufficient to induce the phenotype. Each individual has two copies of the gene, referred to as alleles, one from each parent. The odds of which of the two alleles from each parent is inherited are by chance alone, namely 50%. Genes are units of heredity that are transmitted independently to the next generation. Two genes separated on different chromosomes sort themselves independently through the process of cross-over between the chromosomes. The greater the distance between two loci, the more likely they are to be separated during genetic transmission. A given disease may be due to multiple mutations in the same gene (allele heterogeneity) or to a single or multiple mutations in two or more genes (locus heterogeneity). It is important to keep in mind, however, that within any one family the gene and the mutation responsible for the disease are the same and only rarely would two genes be transmitted for the same disease. Mutations involving only a single nucleotide are known as point mutations and are responsible for 70% or more of all adult single gene disorders (Table 22.1). A point mutation may be due to substitution of one nucleotide for another (missense mutation) or it may change the amino acid to a stop signal (nonsense), which will truncate the protein (truncated mutant); or it may eliminate a stop signal so the protein is elongated (elongated mutant). Nucleotides may be deleted or added which will result in a different reading from left to right and the gene may be read entirely differently, resulting in a non-functioning product (frameshift).
Table 22.1 Cardiac diseases with an identified genetic locus or gene
| Cardiomyopathies | Chromosomal locus |
| Hypertrophic cardiomyopathy | 1q32, 3p21, 11q11, 12q23, 14q12, 15q22, 19p13, 15q11, 14q1, 4q26–27 |
| Dilated cardiomyopathy without conduction defects | 1q32, 6q1, 9q12, 10q24, 15q1, 2q31 |
| Dilated cardiomyopathy with conduction defects | 1q1, 3p22, 6q23 |
| Arrhythmogenic right ventricular cardiomyopathy | 1q12, 2q32, 14q12, 14q23, 3p23, 1q42 |
| Mitochondrial cardiomyopathies | Mitochondrial DNA |
| Cardiac septal defects | |
| Holt–Oram syndrome | 12q2 |
| Di George syndrome | 22q |
| Noonan syndrome | 12q |
| Aortic diseases | |
| Aneurysms | 11q23–24 |
| Supravalvular aortic disease | 9q |
| Marfan syndrome | 15q |
| Conduction disorder | |
| Familial heart block | 19q13, 1q32 |
| Ventricular arrhythmias | |
| Long QT syndrome | 3p21, 4q25, 7q35, 11p15, 21q22, 17q23 |
| Brugada syndrome | 3p21 |
| Idiopathic VT | 3p21 |
Patterns of inheritance of single gene disorders
Autosomal dominant disorders are so named because the disease occurs despite a mutation in only one of the alleles. Males and females are equally affected with about 50% of the offspring expected to have the defective gene (Fig. 22.1). The following features are characteristic of autosomal dominant inheritance:
Figure 22.1 Mendellian patterns of inheritance. Indicates carrier, unaffected and affected patterns of inheritance.
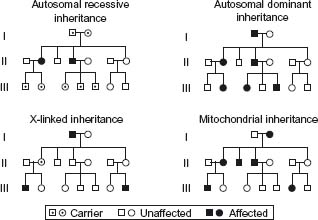
- each affected individual has at least one affected parent
- 50% of the offspring will have the defective gene
- normal children of an affected individual bear only normal offspring
- males and females are equally affected
- both sexes are equally likely to transmit the abnormal allele to male and female offspring and therefore, male-to-male transmission occurs
- vertical transmission through successive generations occurs
- it is typical for autosomal dominant disorders to have delayed age of onset and variable clinical expression.
Autosomal dominance is the predominant form of inheritance in adult cardiovascular disorders and examples would be FHCM and long QT syndrome.
Autosomal recessive inheritance, in contrast, requires both alleles to be defective and thus both parents must have the defective gene. The following features are characteristic of autosomal recessive inheritance:
- generally the parents are clinically normal heterozygotes
- alternate generations are affected with no vertical transmission
- both sexes are affected with equal frequency
- each offspring of heterozygous carriers has a 25% chance of being affected, a 50% chance of being an unaffected carrier and a 25% chance of inheriting only normal alleles.
If one parent is homozygous and the other is heterozygous, the pattern is changed, with 50% being affected and 50% being carriers.
Examples of autosomal recessive disorders affecting the heart include Jervell and Lang-Nielson long QT syndrome and Pompe’s disease.
X-linked inherited disorders are caused by genes located on the X chromosome. Since a female has two X chromosomes, she may carry either one mutant allele or two mutant alleles; the trait may therefore display dominant or recessive expression. Since males have only a single X chromosome, they are likely to display the full syndrome whenever they inherit the abnormal gene from their mother. Hence, the terms X-linked dominant and X-linked recessive apply only to the expression of the gene in females. Since males must pass on their Y chromosome to all male offspring, they cannot pass on mutant X alleles to their sons; therefore, no male-to-male transmission of X-linked disorders can occur. All females receiving a mutant X chromosome are thus carriers and those who become affected clinically are usually homozygous for the defective gene. The characteristic features of X-linked inheritance are as follows:
- no male-to-male transmission
- all daughters of affected males are carriers
- sons of carrier females have a 50% risk of being affected and daughters have a 50% chance of being carriers
- affected homozygous females occur only when an affected male and a carrier female have children
- the pedigree pattern in X-linked recessive traits tends to be oblique because of the occurrence of the trait in the sons of normal carrier sisters of affected males.
Examples of X-linked disorders of the heart include X-linked cardiomyopathy, Barth’s syndrome and muscular dystrophy.
Another uncommon inheritance pattern is that of mitochondrial abnormalities. The mitochondrion has its own genome of about 37 genes contained in 16 K of DNA in a single circular chromosome. Most of the disorders involve oxidative phosphorylation and are usually evident very early in life. Phenotypes due to mitochondrial DNA mutations are transmitted by maternal inheritance only since the ovum has mitochondria but the sperm does not. The characteristic features of mitochondrial inheritance of disease include:
- equal frequency and severity of disease for each sex
- transmission through females only, with offspring of affected males being unaffected
- all offspring of affected females may be affected
- extreme variability of expression of disease within a family
- phenotypes may be age dependent
- organ mosaicism is common.
An example of mitochondrial inherited cardiac disease is the cardiomyopathy of Kearns –Sayre syndrome.
Family history and inherited cardiovascular disorders
Identification of a disease segregating in a particular family is determined from the family history. Obtaining a careful family history has not hitherto been an emphasis for the cardiologist. Recognizing the importance of family history in single gene disorders and also in family cluster disorders such as atherosclerosis and hypertension must be a foremost part of the history and physical examination. Certain ethnic groups may direct specific testing such as for hemoglobinopathies in populations from the Mediterranean or sickle cell disease in the African American. The first individual to be recognized to have the disease is usually referred to as the proband. Once a proband is recognized, information should be collected on all first-, second-or third-degree relatives. The information should include medical problems, pregnancies and information on those individuals related to the family who are deceased. Frequently, it is important to pursue miscarriages, birth defects and other factors that may appear to be unrelated. A pedigree should be constructed analogous to those shown in Figure 23.1, to determine the pattern of inheritance.
Once it has been established that there is a familial disease, it is important to provide information to the individual or parents appropriate to the education of the individual. Every attempt must be made to explain the disease so that important issues are understood by the individual. An attempt must be made to outline the diagnosis, prognosis if known, and mode of transmission together with a discussion of the psychologic and social issues. It is also important in young couples to emphasize the mode of transmission and their chances of passing on the disease as well as the availability of prenatal diagnosis if appropriate. The information must be provided in a non-judgmental and unbiased manner. The family must be able to make a decision with respect to their religion, social and cultural backgrounds. It is sometimes frustrating for the counselor but personal bias must be avoided. Sometimes the issues are extremely sensitive and the options must be presented with concern and compassion but still remaining non-directional.
Single gene cardiovascular disorders
These diseases cover a wide spectrum from structural defects such as familial atrial septum to functional defects such as long QT syndrome (see Table 22.1). For most of these diseases, the chromosomal location (locus) has been mapped but the gene has not yet been identified. However, diseases such as the cardiomyopathies, particularly FHCM, have undergone major investigations with elucidation of the pathogenesis. Animal models of human FHCM have been developed and therapies have been evaluated. There is considerable progress in the identification of genes responsible for ventricular arrhythmias, particularly the long QT and Brugada syndromes. It is still premature to manage these disorders based on their genetic etiology. This is in part because genetic screening is not available and the populations studied are not yet adequately characterized to provide generalized approaches to treatment. A few of these disorders will be discussed to indicate our progress in improving diagnosis, prevention and treatment. The discussion also indicates the trends for the future when most of these genes will be identified and data become available on the pathogenesis and prognosis as they relate to the specific molecular defects.
Long QT syndrome
Several mutations have been identified in cardiac ion channels (sodium, potassium and calcium channel genes) responsible for long QT syndrome (LQT) which predisposes to ventricular arrhythmias and sudden death. Since the initial locus identified was on chromosome 11, seven loci have been mapped and six genes identified (see Table 22.1). Several mutations have been identified in the sodium channel gene SCN5A.10–12 The long QT-associated mutations in SCN5A are associated with increased sodium flux and prolonged depolarization. The mechanism for the arrhythmias is believed to be an imbalance between the inward and outward currents during the plateau of the action potential. Most of the mutations in the sodium channel appear to be gain of function. The pattern of inheritance is most frequently autosomal dominant although a rare recessive form has also been identified.
Several mutations have also been noted in potassium channels, which decrease potassium flux through a loss of function.11,12 These mutations appear to have a dominant negative effect. Rarely, the QT syndrome is inherited in an autosomal recessive manner and may be associated with deafness (e.g. the Jervell and Lang – Nielsen syndromes). This observation led to the recognition that the inward potassium current is necessary for endolymph production in the inner ear.13 There is extensive phenotypic variability among these various genes and mutations and within the same family there are many modifiers yet to be recognized to properly interpret genotype/phenotype correlations.
Mutations are claimed to have some prognostic significance. In general, patients with LQT1 due to the KVLQT1 gene and LQT2 due to HERG have a higher risk of cardiac events than patients with LQT3 due to SCN5A.14 The latter, despite causing fewer events, has a relatively higher mortality, which indicates higher lethality of the events. The first line of therapy in patients with LQT1 is beta-blockers. Preliminary data suggest LQT3 patients might benefit from Na+1 channel blockers, such as mexiletine, but long-term data are not available yet (Class IIb).
A new syndrome has been described associated with shortening of the QT interval on the ECG and clinically with syncope and sudden death. This syndrome, referred to as the short QT syndrome, is due to mutations in the SCN5A gene.15
Another form of cardiac channelopathy is idiopathic ventricular fibrillation. The ECG may be normal although some individuals have an associated electrocardiographic abnormality, including ST segment elevation in V1 – 3 together with right bundle branch block, referred to as Brugada syndrome.16–18 Mutations responsible for this disease have been linked to SCN5A with dominant inheritance. There is at present no proven mechanism for the ventricular arrhythmia but it is believed to be due to heterogeneity between the epicardium and endocardium during repolarization which leads to re-entry.
Brugada syndrome
The ECG pattern is characteristic of this syndrome, consisting of right bundle branch block and ST segment elevation in V1 – 3 and sudden death at a young age.19
SCN5A was identified as the first and, thus far, the only causal gene for Brugada syndrome in 1998.20 Over 60 different mutations in SCN5A have been identified that collectively account for approximately 25% of all cases with Brugada syndrome. As in many other genetic disorders, Brugada syndrome also exhibits locus heterogeneity, and a second locus on chromosome 3 has been mapped.21 However, the causal gene has not yet been identified. The only treatment is an implantable cardioverter-defibrillator (ICD). Once a case is identified, it is recommended the family be screened for the mutation. SCN5A mutations induce a large spectrum of phenotypes, including Brugada syndrome, long QT syndrome (LQT3), isolated progressive cardiac conduction defect, idiopathic ventricular fibrillation, and sudden unexpected death syndrome (SUDS).22
Familial a trial fibrillation
Atrial fibrillation is the most common cardiac arrhythmia with a prevalence of 6% over the age of 65 years.23 The chance of developing atrial fibrillation over age 40 years is about 25% in men and women.24 Atrial fibrillation accounts for about one-third of all strokes and 30% of all patients with atrial fibrillation have a family history of the disease.25 In 1997, Brugada et al identified the first locus on chromosome 10q22 – 24 in a Spanish family.26 Since that time, seven loci have been mapped and five genes identified: KCNQ1, KCNE2, KCNE3, KCNJ2, KCNA5 (Table 22.2). All of the genes identified encode a potassium channel subunit. The mechanism of action by which these genes induce atrial fibrillation is shortening of the action potential duration and shortening of the atrial effective refractory period. The consistency of the mechanism of action suggests the development of therapy specifically targeted to prevent these molecular events.
The most recent finding is that of somatic mutations in the connexin 40 gene.27 This gene (CJA5) encodes for the gap junction protein connexin 40, which is involved in electrical conduction in the myocardium. The mutation was identified through genetic screening of human atrial samples obtained from patients with atrial fibrillation undergoing cardiac surgery for valvular or other diseases. In addition to monogenic diseases, there is the observation that patients with structural heart disease are predisposed to atrial fibril-lation by inherited DNA polymorphisms.28 The recent development of chips with hundreds of thousands of SNPs, enabling genome-wide scans will, over the next few years, elucidate those SNPs that predispose to atrial fibrillation. Within the next decade, one can expect most of the genes responsible for atrial fibrillation and those SNPs that confer predisposition to be identified and therapies will be developed based on an individual’s genomic profile.
Wolf–Parkinson-White syndrome
A gene responsible for an uncommon form of Wolff – Parkinson-White syndrome (WPW) was shown to be AMPK.29,30 Several mutations in AMPK have since been identified31–33 as inducing WPW which is associated with hypertrophic cardiomyopathy and conduction disorders along with a high incidence of atrial fibrillation. It appears that AMPK induces abnormalities in glycogen which leads to all three phenotypes: WPW, conduction disturbance and hypertrophic cardiomyopathy. A genetic animal model of WPW expressing the AMPK gene has been developed which should elucidate pathogenesis and suggest potential therapies.34
Familial hypertrophic cardiomyopathy (HCM)
Clinical and pathologic features of HCM
HCM is an autosomal dominant disease defined by cardiac hypertrophy in the absence of an increased external load (primary hypertrophy). Patients with HCM exhibit protean clinical manifestations ranging from minimal or no symptoms to severe heart failure and sudden cardiac death (SCD). The majority of patients with HCM are asymptomatic or mildly symptomatic. The clinical manifestations often do not develop until the third or fourth decades of life. HCM is a relatively benign disease with an estimated annual mortality rate of < 0.7% in the adult population.35 However, SCD is often the first and tragic manifestation of HCM in the young.36 HCM is the most common cause of SCD in young competitive athletes, accounting for approximately one-third of all SCD.36
The main pathologic features of HCM include myocyte hypertrophy and disarray, interstitial fibrosis and, to a lesser extent, thickening of the media of intramural coronary arteries. While hypertrophy and fibrosis are the common responses of the heart to all forms of injury, myocyte disarray is considered the pathologic hallmark of HCM.37 Cardiac hypertrophy and interstitial fibrosis are the major determinants of mortality and morbidity in HCM.38–40 In those with mild or no cardiac hypertrophy, myocyte disarray is associated with the risk of SCD.41
Molecular genetics
HCM is a genetic disease with an autosomal dominant mode of inheritance. Family history of HCM is present in approximately half of all index cases (familial HCM) and the remainder are sporadic. Sporadic means offspring of parents of whom neither has the mutant gene or the disease. Presumably, the mother was exposed shortly after conception (12 – 24 hours) to a mutagen (EG radiation) that induced in the embryo a mutation that causes HCM. The mutation, albeit de novo, in the affected offspring will transmit the mutation and disease to their offspring in the same pattern as familial. HCM usually is due to mutations in any of at least 12 contractile sarcomeric proteins (Table 22.3). Recently, a new gene (MY0Z2 or myozenin 2) was identi-fied as being responsible for HCM.42 The gene encodes a Z disk protein and thus expands the spectrum of the causal genes to include those encoding the Z disk proteins. Over 500 mutations in 12 genes have been identified.9
Genotype/phenotype correlations
Genotype/phenotype correlation studies suggest that cau sal mutations affect the magnitude of cardiac hypertrophy and the risk of SCD (Fig. 22.2). Mutations in the beta-myosin heavy chain (MyHC) are generally associated with an early onset and more extensive hypertrophy and a higher incidence of SCD.43,44 In contrast, mutations in the myosin-binding protein C (MyBP-C) are associated with a low penetrance, relatively mild hypertrophy, late onset of clinical manifestations and a low incidence of SCD.44–46 Mutations in cTnT are usually associated with a mild degree of hypertrophy but more extensive disarray and a high incidence of SCD.41,47,48 Mutations in alpha-tropomyosin are generally associated with a benign phenotype and mild left ventricular hypertrophy. However, a phenotype of mild hypertrophy and high incidence of SCD also has been described.49 Mutations in essential and regulatory myosin light chains have been associated with mid-cavity obstruction in HCM and skeletal myopathy in some50 but not in others.51 Mutations in titin52 and alpha-actin53–55 have been observed in a small number of families.
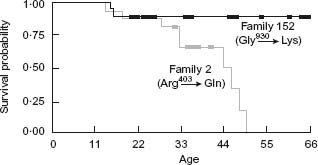
Figure 22.2 Stratification of risk according to mutation. Shown here are two different mutations in the βMHC gene. The mutation in Family 152 is associated with essentially normal lifespan while Family 2 has a mean lifespan of about 28 years. This emphasizes the potential prognostic significance of individual mutations.
Despite the observed associations between severity of the phenotype and the causal mutations, there is considerable variability in phenotypic expression of HCM among affected members, even among those who have identical causal mutations. Overall, the results of genotype/phenotype correlation studies have been subject to a large number of confounding factors, such as the small size of the families, small number of families with identical mutations due to low frequency of each mutation, influence of modifier genes,56 influence of non-genetic factors and rarely homo-zygosity for causal mutations and compound mutations.20,57,58 Correlations in the small number of patients studied suggest prognostic stratification by the mutations but caution must be exercised until larger studies are performed (see Fig. 22.2).
Pathogenesis of HCM
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


