Molecular and Cellular Mechanisms of Myocardial Ischemia-Reperfusion Injury: Introduction
Normal cardiac function is predicated on a continuous supply of oxygen and nutritive substances. When coronary perfusion is interrupted, profound myocardial damage can occur at both microscopic and macroscopic levels. Clinically, this scenario gives rise to acute coronary syndromes manifesting as angina, or in the most severe form, acute myocardial infarction. The onset of ischemia triggers homeostatic processes geared at limiting damage but that may act in concert with processes associated with reperfusion to actually exacerbate injury. These potentially deleterious effects of reperfusion have been described as reperfusion injury, or more aptly, ischemia-reperfusion injury, because the pathology associated with reperfusion occurs only in the setting of antecedent ischemia. Ischemia-reperfusion injury is a complex process that is brought about by the interaction of a number of cells, including endothelium and inflammatory cells, with components of the coagulation and complement cascades. This interplay promotes the formation of harmful substances, which may further amplify myocardial cell death after an ischemic insult.
Pathophysiology of Myocardial Ischemia
Myocardial ischemia occurs when there is a demand-supply mismatch between the myocardium’s energy requirement and the oxygen and other substrate supply delivered via myocardial perfusion.1 With compromise of coronary arterial flow, a number of electrical, mechanical, and chemical changes take place in the ischemic area of myocardium. The myocardium becomes cyanotic with consumption of freely diffusible oxygen and oxymyoglobin oxygen stores causing decreased tissue oxygen tension.2 With increasing tissue hypoxia, intracellular respiration shifts from its aerobic to its anaerobic form. Adenosine triphosphate (ATP) stores are rapidly depleted,3 causing adenosine diphosphate (ADP), adenosine monophosphate (AMP), and adenosine to accumulate in the tissue. At this point, the ischemic region of myocardium loses its ability to maintain its normal negative resting membrane potential.4 Diminished ATP stores and/or alteration in availability of calcium5 lead to cessation of cardiac contraction.4 This is followed by a distension of the ischemic myocardium, which perhaps occurs as the result of stretch due to tugging by adjacent nonischemic (and still contracting) muscle.6 Characteristic metabolic changes occur in the ischemic tissue, including an accumulation of tissue lactate,3,7 H+ ions,8 phosphate,7 and potassium.9,10 There is also an increase in tissue tension of carbon dioxide (Pco2),11 as an accumulating by-product of cellular metabolism in the absence of an egress mechanism during the stasis that characterizes ischemia. In addition, mitochondrial calcium increases,12 a process that may further contribute to ischemic contracture and perhaps even the ultimate death of the vulnerable myocyte. In an autoregulatory attempt to increase blood flow, arterioles exhibit a profound vasodilator response,11 which under nonobstructive conditions might result in a restoration of nutritive flow; however, under severe ischemic conditions, it is often futile. If complete obstruction to blood flow persists for as little as 20 minutes, myocardial necrosis may be observed.13 Under circumstances in which myocardial reperfusion is re-established with great rapidity, however, mechanical function can return to near baseline levels.14
When ischemic myocardial tissue is subjected to detailed histopathologic examination, characteristic structural changes can be seen, one of the earliest of which includes a decrease in the size and number of glycogen granules within the myocytes. The amount of glycogen decreases significantly after 30 minutes of ischemia and almost disappears during the subsequent 2.5 hours.15-17 Myofibrils then appear to arrest in a relaxed state.18 Ischemic tissue shows marked enlargement of interfibrillar and subsarcolemmal spaces with swelling of transverse tubules (T-tubules). Mitochondria also appear swollen and demonstrate decreased matrix density.15-17 This swelling reflects changes in intracellular fluid distribution and increases in myocardial water content, which can represent nearly a 50% increase after an extended period of interrupted blood flow.19,20 In addition to the contribution of intracellular shifts of fluid, there is an expansion of the extracellular water content, presumably brought about by increases in vascular permeability in vessels in the ischemic zone.21-23
There remains considerable controversy in the field as to the precise point in this complex process at which the death of cardiac myocytes becomes irreversible. There are many pathologic mechanisms triggered that, if remained unchecked, can lead to the inevitable death of the myocyte. However, other death-sparing pathways may be simultaneously activated and counterbalance the prodeath signals. The ultimate fate of the cardiac myocyte, therefore, is determined by the prevailing balance of forces promoting or inhibiting its death. Clinically, it is often difficult to know where this balance lies or whether a given therapy will hasten or salvage myocyte death. An ischemic insult can be considered to cause irreversible myocyte death if it is of sufficient magnitude and duration so that cells continue their march toward death even after restoration of blood supply.24 Dead myocytes exhibit a characteristic histologic appearance, including the presence of contraction bands25-27 and amorphous densities within swollen mitochondria.27 Irreversible damage to mitochondria and cell membranes causes the release into blood of numerous intracytoplasmic proteins that can be detected in plasma to diagnostically quantify the degree of myocardial necrosis in patients and that may signal the primitive “danger” response that triggers immune activation. Markers of cardiac myocyte death appear in the plasma as plasma membrane integrity is compromised; these include enzymes such as lactate dehydrogenate, creatinine phosphokinase (CK), serum glutamic oxaloacetic transaminase, and other intracellular proteins, such as myoglobin, troponins T and I, and cardiac-specific myosin light chains.28 The other factor that contributes to rapid washout of these markers into blood is reperfusion of myocardium following the period of ischemia. Integration of the area under the time-activity curves provides a rough estimate of extent and degree of infarction,29 although the presence of reperfusion causes early washout peaks that can somewhat confound quantitative interpretation of this information. There is now greater appreciation of the role of inflammation, vascular remodeling, and plaque vulnerability in modulating ischemic injury in acute coronary syndromes. Recent literature points to other potential markers of adverse outcomes that include C-reactive protein,30 N-terminal pro-brain natriuretic peptide,31 soluble CD40 ligand,32,33 myeloperoxidase,34 metalloproteinases,35 tissue inhibitors of metalloproteinases,36 and adiponectin.37
Pathophysiology of Myocardial Ischemia-Reperfusion Injury
Restoration of myocardial blood flow following a period of flow interruption, called reperfusion, is beneficial in that it arrests the advancing wavefront of myocyte death. Conversely, it also brings in cellular and humoral factors into a primed milieu, setting the stage for potential coagulation events that impede reflow, complement-mediated tissue destruction, local generation of damaging reactive oxygen and nitrogen species, and incursion of cells that can further obstruct flow or promote inflammation. An example of an ischemic and nonreperfused myocardium, as well as an ischemic myocardium that had blood flow restored, is shown in Fig. 58–1. These processes of coagulation and inflammation initiate a cascade of reparative events that can lead to fibrosis in previously frankly necrotic zones (Fig. 58–2). Understanding the time frame and cellular processes are important for the understanding of the postischemic histopathology and pathophysiology of the healing infarct, with clinical implications of myocardial scar formation, impaired systolic/diastolic function, aneurysm formation, or cardiac rupture.
Figure 58–1

Reperfusion of ischemic myocardium results in a decreased area of infarction. Murine myocardium infarcted using permanent coronary artery occlusion (left) is compared with myocardium reperfused after 3 hours of occlusion (right). Viable tissue stains brick-red with triphenyltetrazolium chloride, indicating that the reperfused tissue has a smaller area of infarct.
Figure 58–2
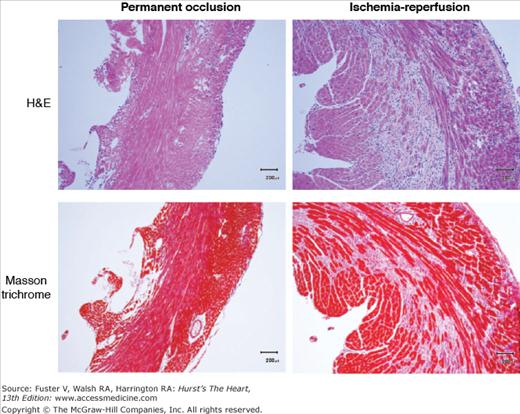
Comparison of macrophage infiltration (top, hematoxylin and eosin [H&E] stain) and collagen deposition (bottom, Masson trichrome stain) in murine myocardium infarcted using permanent coronary artery occlusion (left) versus 1 hour of occlusion followed by reperfusion. The reperfused myocardium shows an increase in both macrophage infiltration and collagen deposition. In each case, the tissue was harvested 3 days after infarction.
Neutrophils (polymorphonuclear leukocytes) are key cellular effectors in the pathogenesis of myocardial ischemic-reperfusion damage. There are compelling data from animal studies showing the protective effects of therapies that either interfere with neutrophil adhesion mechanisms or compromise neutrophil function prior to ischemia and reperfusion.38-40 A number of events must occur for recruitment of the requisite quantity of the neutrophils to elicit myocardial damage in the setting of ischemia-reperfusion. Neutrophils are activated and recruited by proinflammatory signals such as tumor necrosis factor-α (TNF-α), complement fragments, and cytokines, which are released by the ischemic-reperfused myocardium itself41-45 or other resident or vascular cells in the vicinity. Intravascular neutrophils decelerate46 and begin to adhere to endothelium primarily through an interaction via P-selectin glycoprotein 1 (PSGL-1) and L-selectin on the neutrophil and P-selectin on the endothelium. PSGL-1 is the most well-characterized ligand for all selectins, and its dual roles include mediation of attachment of leukocytes to the activated endothelium47-49 and signal transduction for leukocyte activation.50-53 Although L-selectin is constitutively expressed on the neutrophil cell surface, P-selectin is stored in α granules of platelets and Weibel-Palade bodies of endothelial cells and is rapidly translocated to the cell surface of these cells within minutes of cardiac reperfusion.54 Hypoxia itself is sufficient to induce endothelial surface expression of P-selectin.55 This interaction brings the neutrophils into close approximation with the vascular wall to engage other cognate receptors, such as L-selectin, platelet activating factor, intercellular adhesion molecule-1 on the endothelial cells and leukocyte β2-integrins (CD11/CD18 complex) on the neutrophils.42 Migration across cell junctions requires interaction between neutrophils and platelet endothelial cell adhesion molecule (PECAM), another immunoglobulin adhesion molecule. Blockage of PECAM using antibody fragments decreases neutrophil accumulation and infarct size in rat myocardium during the reperfusion that follows ischemia.56
Infiltration of neutrophils into the ischemic zone begins within minutes after the onset of ischemia and increases significantly for up to 90 minutes after reperfusion. A canine model comparing permanent occlusion with reperfusion after temporary occlusion showed that accumulation of neutrophils in myocardium was augmented by 80% in reperfused infarcts, likely due to reperfusion-facilitated neutrophil entry into necrotic regions.57 Stimulation of neutrophils by chemotactic factors and cytokines elicits the respiratory burst characterized by sudden release of oxygen-derived free radicals including superoxide anion, hypochlorous acid, hydroxyl radical, chloramines, and hydrogen peroxide.42 This cytotoxic milieu damages endothelial cells and impairs physiologic vasodilatory and anticoagulant mechanisms. These events promote increases in vascular permeability, leukocyte infiltration, and neutrophil and platelet activation and adhesion, thereby further amplifying reactive oxygen species (ROS) release and exacerbating reperfusion injury.
Resident and recruited macrophages also play a role in early reperfusion injury in that they are sources of proinflammatory cytokines including interleukin (IL)-1β,58 TNF-α,59 and IL-6.60 Together, these cytokines function to attract and activate other leukocytes.
The intact endothelium that lines blood vessels maintains an antithrombotic phenotype to maintain blood fluidity. Normally the luminal surface of vascular endothelium repels platelets and coagulant events and prevents contact of the coagulation system with the subjacent and highly procoagulant subendothelial matrix (which is rich in collagen and tissue factor).61 The endothelial surface is rich in heparin-like proteoglycans that can function to accelerate the inactivation of coagulation proteases by antithrombin III. Another mechanism by which prevention of thrombus formation is achieved involves the membrane-spanning thrombin-binding protein, thrombomodulin, which converts thrombin into a potent protein C activator.62 In addition, local production of nitric oxide potently inhibits platelet aggregation and promotes local vasodilatation.
Under conditions associated with tissue hypoxia such as in myocardial ischemia, the endothelium develops a prothrombotic phenotype. Macrophage, monocyte, and endothelial cell expression of tissue factor increases, whereas endothelial cell expression of thrombomodulin is significantly reduced.63-66 Moreover, retraction of endothelial cell margins exposes the procoagulant subendothelial matrix to circulating elements of the coagulation cascade. Additionally, hypoxia triggers the calcium-dependent exocytosis of Weibel-Palade bodies, leading to increased secretion of von Willebrand factor.67 Platelet adhesion and aggregation are fostered by the superoxide-mediated decline in NO levels that occurs with reperfusion,68-70 and this also contributes to a local prothrombotic diathesis. Platelets activated by ischemia-reperfusion not only aggregate and cause microvascular plugging, but also release products such as thromboxane and serotonin that give further rise to microvascular inflammation and local vasospasm.71 There is also some evidence that activation of platelets, endothelium, and leukocytes can release microparticles that either signal downstream cells or that can contribute to inflammation and anticoagulation at sites remote from the primary ischemic one.
Because of their key roles as metabolic energy producers and regulators of apoptosis, mitochondria play a central role in cell survival.72 Mitochondrial injury is an important component in the pathobiology of ischemia-reperfusion injury. Arguably the most critical factor in determining mitochondrial injury is opening of the permeability transition pores (PTPs), large proteins that form nonselective pores in the inner mitochondrial membrane. PTP opening leads to immediate depolarization of the mitochondrial membrane and transforms the mitochondria from net ATP producers to net ATP consumers by reversing ATP synthesis in an attempt to maintain the membrane potential. PTPs are believed to be sensitive to calcium, redox, voltage, and pH states.73 Ischemia serves to prime the mitochondrial permeability transition (MPT), whereas reperfusion triggers the process. Under ischemic conditions, activated fatty acids accumulate in the myocardium, which not only decreases the rate of ATP synthesis, but also increases both inner mitochondrial membrane leakiness and cytochrome c mobilization and release.74 This may be an important ischemia-reperfusion–mediated event given the pivotal role this could play as an apoptotic trigger. During ischemia, acidosis, elevated magnesium levels, and diminished electron transport generally keep PTPs closed; however, with reperfusion, mitochondria are placed at high risk of PTP opening due to the associated ROS burst and clearing of acidosis, as well as the elevated calcium and inorganic phosphate levels.73 Recent work has shown that vagal nerve stimulation and subsequent acetylcholine release can inhibit MPT opening, resulting in protection from reperfusion injury.75
Cytosolic calcium overload is believed to be an important mechanism underlying development of ischemic contracture and cardiomyocyte death.76,77 Whereas ischemia disrupts the myocyte’s capacity to regulate calcium, it is reperfusion that dramatically leads to intracellular accumulation of toxic levels of calcium.78,79 There are several potential mechanisms for this phenomenon.80 One is related to increased sarcolemmal calcium entry via L-type calcium channels that occurs as a consequence of sodium overload.81,82 Another possibility links increased myocyte entry of calcium to a perturbation in calcium handling by the sarcoplasmic reticulum secondary to ATP depletion–related disruption of the sarcolemmal membrane.80 Additionally, there is evidence to suggest that reperfusion injury causes significant myofilament desensitization to calcium. Moreover, in the minutes following reperfusion, intracellular calcium overload and depletion of ATP levels give rise to hypercontracture or sustained shortening and stiffening, which can lead to myocardial tissue necrosis. Opening of mitochondrial83 and/or sarcolemmal84 ATP-sensitive potassium channels (KATP) has been shown to decrease the extent of ischemia-reperfusion injury, in part by reducing calcium overload, and is a topic of active research.
Toll-like receptors (TLRs) are transmembrane pattern recognition receptors that recognize pathogen-associated molecular patterns indicative of invading microbes. Activated TLR signaling results in the release of a wide variety of interferons, cytokines, and chemokines that prime the innate and adaptive immune responses. The robust leukocyte infiltration and activation that are central to ischemia-reperfusion injury has led to considerable interest in understanding the potential pathologic roles played by TLR2 and TLR4. Elegant experiments with murine bone marrow chimeras have shown that TLR2 deficiency on both leukocytes and non–bone marrow–derived cells results in protection during reperfusion, as evidenced by smaller myocardial infarct size, reduced ROS production, and less leukocyte infiltration.85 TLR4-deficient mice exhibit smaller myocardial infarct sizes and decreased peri-infarct inflammatory mediators,86 and pretreatment with a TLR4 antagonist results in a smaller area of infarct after transient left anterior descending artery occlusion in mice.87
With reperfusion of previously ischemic myocardium, the reintroduction of abundant oxygen immediately evokes a burst of potent oxygen-derived free radicals, such as superoxide anion, hydroxyl radical, hydrogen peroxide, and peroxynitrite.88-90 These ROSs are generated by endothelial cells, recruited/activated leukocytes, and even the myocytes themselves. They are, in most instances, the by-product of mitochondrial respiration, catecholamine oxidation, the Haber-Weiss pathway, and the enzyme activities of xanthine oxidase, cytochrome oxidase, and cyclooxygenase.91-96 The overwhelming increase in oxygen-derived free radicals can peak within minutes but may persist for many hours after restoration of blood flow. This overwhelms the tissue’s antioxidant capacity, leading to protein denaturation, inactivation of key homeostatic enzymes, and peroxidation of lipid membranes.97-99 These processes cause loss of membrane integrity, ultimately giving rise to necrosis and cell death. There are several lines of evidence confirming the in vivo formation of ROS in the human heart.
The specific role of nitric oxide (NO) in myocardial damage and ischemia-reperfusion injury remains controversial. To the extent that endothelial injury and dysregulation of microvascular tone are features of myocardial ischemia-reperfusion injury, it is hardly surprising that NO has been implicated in this phenomenon.100 NO causes relaxation of vascular smooth muscle by activating intracellular guanylate cyclase, causing an increase in cyclic guanosine monophosphate levels.101 In addition to its vasodilatory function, NO has other important vascular functions as well, such as inhibition of platelet adhesion and aggregation, inhibition of neutrophil adhesion, and maintenance of endothelial restricted diffusion.102-105 NO may have dual roles in determining cell survival in myocardial ischemia-reperfusion injury. NO can inhibit apoptotic death of myocytes via nitrosylation of active sites of caspases.106 Alternatively, high NO levels may directly contribute to myocyte apoptosis. Although the synthesis of NO is ongoing during the ischemic phase, reoxygenation triggers a precipitous decrease in NO, and this decline is precipitated by the quenching effect of ROS generated during reperfusion.100,107 Several studies have demonstrated that administration of NO donors or induction/overexpression of the enzyme responsible for synthesis of NO may potentially prevent myocardial ischemia-reperfusion injury.103,108-111 Curiously, there is equally compelling evidence that pharmacologic inhibition of NO production may also reduce infarct size, probably by preventing peroxynitrite formation from NO and superoxide during reperfusion.112-115
Activation of the complement system has been implicated in ischemia-reperfusion injury.116 Activation of complement by either the classical, alternate, or mannose binding pathways causes cleavage of the C3 component, thus causing formation of the anaphylatoxins (C3a and C5a) and the membrane attack complex (C5b-9, MAC). The MAC is the final effector that punctures the target cell causing rapid dissipation of ionic gradients and extrusion of cytoplasmic contents. Complement activation elicits myocardial damage in ischemia-reperfusion injury via two mechanisms. First, the anaphylatoxins (C3a and C5a) and chemotactic factor C4a augment superoxide production and cellular release of histamine and platelet-activating factor, thereby increasing vascular permeability and attracting neutrophils to the ischemic area.116 Even small amounts of C5b-9, below the threshold levels required for cell lysis, may trigger translocation of P-selectin to the endothelial surface, further contributing to neutrophil adhesion. Second, activation of the complement system also causes direct myocardial damage through the formation of the MAC. MAC has been detected using immunohistochemical techniques in areas of myocardial infarction with relatively little deposition in adjacent nonischemic regions.117 In experimental models, there is a plethora of evidence indicating that local activation of the complement system is harmful to the heart. In one model, depletion of complement by the administration of cobra venom toxin significantly reduced ischemic injury,118,119 whereas in other models, soluble complement inhibitors (sCR-1) and a C1 esterase inhibitor have been used to eliminate the deleterious effect of complement cascade activation on myocardium.116,120,121
Apoptosis has been noted to increase during the reperfusion of myocardial ischemic injury.122 Given the multiple, complex mechanisms at work in the ischemic milieu, it is helpful to think of the process of accelerated apoptosis as an increase in the ratio of proapoptotic to antiapoptotic mechanisms. Two such opposing processes are triggered by the proteins bcl-2 (antiapoptotic) and Bax (proapoptotic).123 In the setting of ischemia and reperfusion, p53 is a transcriptional regulator that helps shift the ratio in favor of apoptosis through direct activation of the Bax gene,124 activation of caspases, and upregulation of the renin-angiotensin system (RAS). The RAS produces angiotensin II, which induces apoptosis directly through increased cytosolic Ca2+ release.125 Furthermore, blockade of the angiotensin II receptor AT1 with quinaprilat decreases p53 and Bax, indicating that the RAS may have an important potentiating effect on p53.122 Binding of angiotensin II to vascular AT1 receptors contributes further to ischemia-reperfusion injury by limiting forward flow, despite removal of the obstruction. Angiotensin II may directly exacerbate microvascular vasoconstriction and promote diastolic dysfunction by boosting intracellular myocyte calcium levels.
Mechanisms of Cardiac Myocyte Death
Myocardial ischemic injury results from a mismatch between myocyte oxygen demand and arterial oxygen delivery. ATP production by mitochondrial oxidative phosphorylation is profoundly dependent on an adequate supply of oxygen, and hypoxemia leads to a rapid decrease in ATP production. Compensatory anaerobic glycolysis produces ATP less efficiently and leads to the accumulation of hydrogen ions and lactate. The resulting acidosis eventually inhibits glycolysis. Without ATP, ion transport mechanisms such as the Na+, K+-ATPase are inhibited, leading to an efflux of K+ and an influx of Na+, Ca2+, Cl–, and H2O. The increase in intracellular Ca2+ augmented by dysregulation of transport systems in the sarcolemma and sarcoplasmic reticulum activates proteases, which alter contractile proteins. Thus, myocyte contractility is impaired despite elevated cytosolic calcium.82 Activation of phospholipase also occurs and results in the release of free fatty acids and lysophospholipids, which incorporate into membranes and further impair function. Free fatty acids such as these are targets of ROSs, which are present in the ischemic milieu. This ATP-independent process of dysregulated cell membrane permeability, accumulation of electrolytes and other pro-oncotic molecules within the myocyte, and resultant cell swelling is termed oncosis, which ultimately results in irreversible injury and cell lysis (Fig. 58–3).
Figure 58–3

Ischemia-induced myocyte death. Obstruction of the arterial lumen by clot leads to gross anatomic and intracellular ischemic changes. The left side of the cell represented in the lower portion of the image shows normal cell function and morphology. The right side of the same cell portrays the major cellular effects of ischemia: free radicals damage the cell wall, mitochondrial dysfunction leads to mitochondrial swelling and release of stored calcium, calcium activates proteases that destroy the cytoskeleton, and dysfunction of ion exchange pumps eliminates vital ion concentration gradients.
Not all myocytes die as the result of cell swelling due to oncosis. Apoptosis is a complex process of programmed cell death that is the result of endogenous or exogenous stimuli and that results in death following cell shrinkage (Fig. 58–4). First described by Kerr et al126 in rat breast carcinomas in 1972 and later by Gottlieb et al127 in rabbit myocytes, apoptosis is an ATP-dependent128 process that culminates in chromosomal DNA fragmentation. The process most commonly involves activation of a family of cysteine aspartyl proteases known as caspases
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


