Chapter 3 Molecular and Cellular Basis of Cardiac Electrophysiology
Basic Concepts
Membrane Potential and Conduction
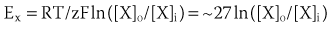
where R is the gas constant, T is temperature, F is Faraday’s constant, and z is the valence of the ionic species. If the resting cardiac myocyte is assumed to have an intracellular [K+] of ~150 mmol/L and an extracellular [K+] of 4 mmol/L, then the Nernst potential for K+ (EK) is roughly –90 mV. The Nernst potential represents the voltage at which the osmotic tendency for K+ to flow across the membrane is exactly balanced by the electrical tendency to flow in the opposite direction, which results in a net zero ionic flux and thus no net current flow. Accordingly, the K+ Nernst potential is sometimes referred to as the zero current potential, at which no K+ current would flow through open K channels. By contrast, the Nernst potential for Na+ in the resting cell is approximately +60 mV, indicating that if a pathway for Na+ to enter the cell were present, Na+ would enter the cell to move the membrane voltage (mV) toward the Nernst potential for Na+. Indeed, this is precisely what happens when Na channels are open and initiate phase 0 of the action potential. The Nernst potential for potassium is, in fact, very close to the resting membrane potential of the ventricular myocyte, indicating that the resting heart cell membrane is highly permeable to K+. Actual resting potentials are less negative than EK due to small conductances of other ionic species with less negative Nernst potentials. In the course of the cardiac cycle, the cell membrane becomes permeable to different ionic species, and these changes in permeability determine time-dependent changes in the membrane potential, with each ion striving to move the membrane voltage to its Nernst potential and inscribing regionally specific action potentials (Figure 3-1).
Passive Membrane Properties and Cable Theory
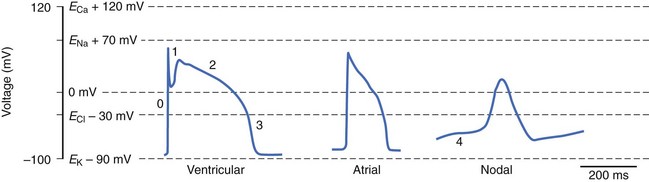
The cardiac cell membrane can be modeled as a circuit comprising variable resistors (ion channels) in parallel with a capacitor (lipid bilayer), an RC circuit (Figure 3-2). The flow of current across the membrane will alter the charge on the capacitor (and therefore the membrane potential) and change the membrane resistance. The flow of current occurs not only across but obviously along the inside and outside of the cell membrane from cell to cell as well, that is, current propagates.
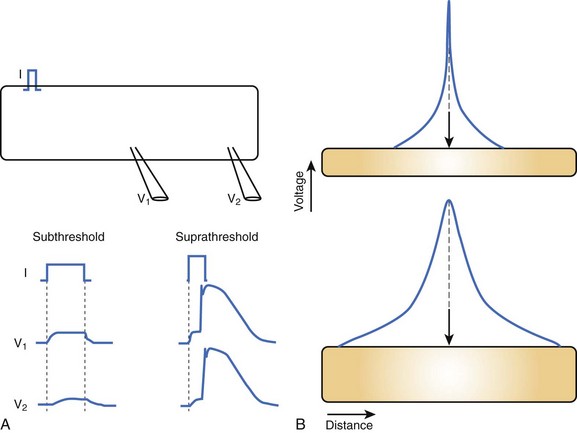
Cable theory, originally developed to understand current flow in trans-oceanic telegraphic cables, can be used to model passive current flow and propagation in a cardiac muscle fiber. In their simplest formulation, the cable equations define the distribution of voltage along a continuous, uniform cable of infinite length stimulated by a point source. The predictions of cable theory are that (1) a change in voltage exhibits a characteristic decay along the cable defined by the space constant (distance over which voltage decays to 1/e of the value at the site of injection) (Figure 3-3, A); and (2) there is an inverse relationship between resistance to current flow (both transmembrane and intercellular) and the cable diameter (Figure 3-3, B). That is, the space constant is directly proportional to the cable diameter, and thus, greater lengths of the cable are influenced by the same current injection into a thick, rather than a thin, cable.
Substantial anatomic and biophysical limitations exist when applying the cable theory description of conduction to cardiac muscle. Anatomically, the shape of the heart is complex, and at any level of integration above a single muscle fiber, it does not resemble a cable. Conduction through the myocardium is not continuous; instead, myocardial cells are connected by gap junction channels that create a non-uniformity of intercellular resistance. Macroscopic discontinuities such as fibrous tissue and blood vessels also significantly perturb the cable view of conduction in the myocardium. Finally, the cardiac cell membrane does not just comprise RC circuits; instead, when stimulated to the threshold, it will generate action potentials (see Figure 3-3, A). Despite the limitations of cable theory, it serves as the foundation for several important concepts in impulse propagation and was used to demonstrate the electrical nature of conduction in the heart.1
While this discussion implicitly treats the activation wave in one dimension, the behavior of propagating waves in the heart is more complex. The complexity can be appreciated if one considers propagation in two dimensions. In this circumstance, the shape of the wavefront is a major determinant of the efficiency of propagation. A convex wavefront, as might be observed after point stimulation, creates a large sink around a smaller activating source. This mismatch reduces conduction velocity and the safety factor for propagation. Conversely, a concave activation front produces a source-sink mismatch that favors the source; this results in a high safety factor and more rapid impulse transmission. Thus, not only do source-sink characteristics influence propagation, they also influence the curvature of the wave front (see Figure 3-4).
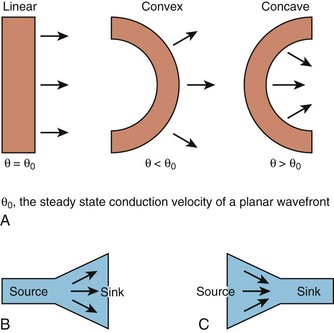
Directionally different conduction velocity is a characteristic feature of cardiac muscle known as anisotropic conduction. Anisotropic conduction has its basis in the structure of the myocyte and cardiac tissue; myocytes are rod shaped and are organized in bundles that are oriented along the long axis of the cell. Transmurally, the axes of these bundles undergo significant changes in orientation through the ventricular wall (~120 degrees maximal deviation).2 Communication among myocytes occurs via gap junction channels that are non-uniformly distributed over the surface of the heart cell, with larger numbers of channel poised to propagate the impulse longitudinally, rather than transversely, to the long axis of the muscle fiber.3 The implications of anisotropy for conduction in the longitudinal and transverse direction under pathologic conditions are controversial. In the context of uniform depression of conduction, as might exist with antiarrhythmic drug treatment, propagation in the transverse direction is preserved compared with conduction in the longitudinal direction. However, when cellular uncoupling occurs, such as in ischemia, longitudinal propagation may exhibit a higher safety factor than does transverse conduction.
Major Breakthroughs: Voltage Clamp, Molecular Cloning
A stimulus of sufficient magnitude applied to a myocyte (or any excitable cell) elicits a typical change in membrane potential known as the action potential. The ionic current basis of the action potential was confirmed and quantitatively studied using the voltage clamp developed in the middle of the twentieth century.4 Voltage clamping is a technique whereby the experimenter controls the transmembrane voltage and measures the current at that defined voltage. Much of what we know about ionic currents in myocytes comes from voltage clamp experiments and a more recently developed type of voltage clamp called the patch clamp.5 A variant of the patch clamp technique permits the measurement of ionic currents through single ion channels.
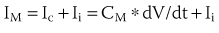
Molecular Basis of Cardiac Action Potentials
Cardiac myocytes possess a characteristically long action potential (200 to 400 ms, see Figure 3-1), compared with neurons or skeletal muscle cells (1 to 5 ms). The action potential profile is sculpted by the orchestrated activity of multiple ionic currents, each with its distinctive time- and voltage-dependent amplitudes. The currents, in turn, are carried by complex transmembrane proteins that passively conduct ions down their electrochemical gradients through selective pores (ion channels), actively transport ions against their electrochemical gradients (pumps, transporters), or electrogenically exchange ionic species (exchangers).
Action potentials in the heart are regionally distinct. The regional variability in cardiac action potentials is the result of differences in the numbers and types of ion channel proteins expressed by different cell types in the heart. Further, unique sets of ionic currents are active in pacemaking and muscle cells, and the relative contributions of these currents may vary in the same cell type in different regions of the heart.6
Ion Channels and Transporters: Molecular Building Blocks of the Action Potential
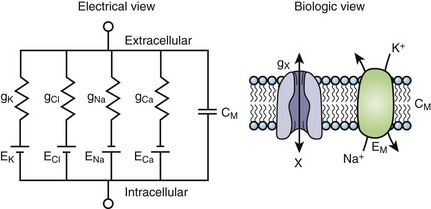
Currents that underlie the action potential are carried by complex, multi-subunit transmembrane glycoproteins called ion channels (Table 3-1). These channels open and close in response to a number of biologic stimuli, including a change in voltage, ligand binding (directly to the channel or to a G-protein–coupled receptor), and mechanical deformation. Other ion-motive transmembrane proteins such as exchangers and transporters make important contributions to cellular excitability in the heart. Ion pumps establish and maintain the ionic gradients across the cell membrane that permit current flow through ion channels. If pumps, transporters, or exchangers are not electrically neutral (e.g., 3 Na+ for 1 Ca2+), they are termed electrogenic and can further influence electrical signaling in the heart.
The most abundant superfamily of ion channels expressed in the heart consists of voltage-gated ion channels. Various structural themes are common to all voltage-dependent ion channels. First, the architecture is modular, consisting either of four homologous subunits (in K channels, see Figures 3-7 and 3-8) or of four internally homologous domains (in Na and Ca channels) (see Figures 3-5 and 3-6). Second, proteins wrap around a central pore (see Figure 3-8). The pore-lining (P segment) regions exhibit exquisite conservation within a given channel family of like selectivity (e.g., jellyfish, eel, fruit fly, and human Na channels have very similar P segments), but not among families with different selectivities. Third, the general strategy for activation gating (opening and closing in response to changes in the membrane voltage) is highly conserved: The fourth transmembrane segment (S4), typically studded with positively charged residues, lies within the membrane field and moves in response to depolarization, thus opening the channel. Fourth, most ion channel complexes include not only the pore-forming proteins (α-subunits) but also auxiliary subunits (e.g., β-subunits) that modify channel function.
Sodium Channels
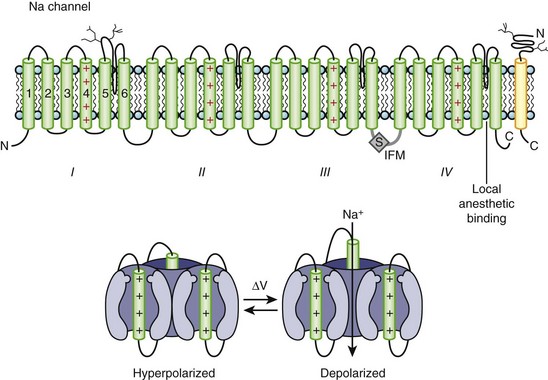
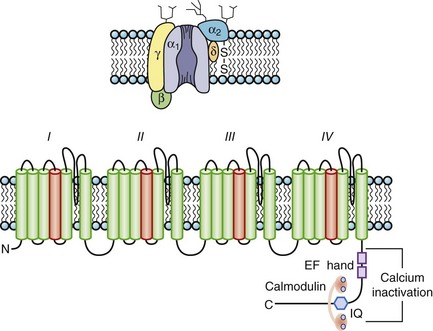
Na channels were the first ion channels to be cloned and have their sequence determined.7 In humans, more than 10 distinct Na channel genes have been cloned from excitable tissues, with striking homology to the complementary DNA (cDNA) cloned from eel electroplax. The cardiac Na channel gene (SCN5A) resides on the short arm of chromosome 3 (3p21) (see Table 3-1). The Na channel complex is composed of several subunits, but only the α-subunit is required for function. Figure 3-5 shows that the α-subunit consists of four internally homologous domains (labeled I to IV), each of which contains six transmembrane segments. The four domains fold together so as to create a central pore, whose structural constituents determine the selectivity and conductance properties of the Na channel.
One of the seminal contributions of Hodgkin and Huxley was the notion that Na channels occupy several “states” (which are now viewed as different conformations of the protein) in the process of opening (activation); yet another set of conformations is entered when the channels close during maintained depolarization (inactivation).4 The m gates that underlie activation and the h gate that mediates inactivation were postulated to have intrinsic voltage dependence and to function independently.8 While some of the implicit structural predictions of that formulation have withstood the test of time, others have not. For example, the four S4 segments are now widely acknowledged to serve as activation voltage “sensors.” In the process of activation, several charged residues in each S4 segment physically traverse the membrane (see Figure 3-5, bottom panel). The contributions of each S4 segment to activation are markedly asymmetrical; some of the charged residues play a much more prominent role than do others in “homologous” positions.9 Other studies have revealed that activation is coupled with inactivation. Indeed, the time course of current decay during maintained depolarization predominantly reflects the voltage dependence of activation, although single-channel inactivation itself does vary with voltage (particularly in cardiac Na channels). If the S4s are the sensors, where are the activation “gates” themselves? This crucial question still remains unresolved. However, according to experimental evidence, S6 is the leading contender for the physical activation gate.
Inactivation of Na channels is as arcane a process as activation. Not only is there loose coupling to activation, but there are multiple inactivation processes. One common approach to distinguishing inactivated states is to determine the rate at which they recover the ability to activate: Repriming from the traditional “fast” inactivation occurs over tens of milliseconds, while recovery from “slow” inactivation may need tens of seconds or longer. Fast inactivation is at least partly mediated by the cytoplasmic linker between domains III and IV (the crucial residues are labeled IFM in Figure 3-5), which may function as a hinged lid, docking onto a receptor formed by amino acids in the S4-S5 linkers of domains III and IV. This notion is consistent with observations that fast inactivation can be disrupted by internal proteases. Nevertheless, it is increasingly clear that mutations scattered widely throughout the channel affect inactivation gating. The structural determinants of slow inactivation are less well localized than those of fast inactivation. Mutations in the P region of domain I affect both activation gating and slow inactivation, while various widely scattered disease mutations identified in paramyotonia congenita and other skeletal myopathies suppress slow inactivation of the Na channel.
Four different β-subunits (NaVβ1 to 4, SCN1B-SCN4B) have been isolated, and all appear to be single membrane–spanning domain (type I topology) proteins with a large extracellular V-shaped immunoglobulin (Ig) fold often found in cell adhesion molecules and small carboxyl terminal cytoplasmic domain.10 The effects of the particular β-subunit on the kinetics and voltage dependence of the α-subunit vary depending on the particular β-subunit and the cell expression system. Despite the expression of β1 mRNA, using subtype-specific antisera, the functional role of β-subunits in cardiac Na currents is still being debated. Several pieces of evidence are consistent with a role for β-subunit(s) in heart cells. First, β-subunits are found in cardiac myocytes, although no NaVβ1 is found in association with the α-subunit protein from the rat heart. Second, NaVβ1 variably modulates the function of NaV1.5 in heterologous expression systems, including the sensitivity of the channel to blockade by antiarrhythmic drugs and free fatty acids. Finally, mutations in β-subunits have been directly or indirectly implicated in heritable cardiac arrhythmias.
In contrast to PKA, protein kinase C (PKC) alters the function of all of the mammalian Na channel isoforms. The PKC effect is largely attributable to phosphorylation of a highly conserved serine in the III-IV linker (see Figure 3-5). Conventional PKC isoforms reduce the maximal conductance of the channels and alter gating in an isoform-specific fashion. The macroscopic current decay of neuronal channels is uniformly slowed by PKC, which suggests destabilization of the inactivated state. Cardiac channels exhibit a hyperpolarizing shift in the steady-state availability curve consistent with an enhancement of inactivation from closed states.
Alteration of ion channel function is an important pathophysiologic mechanism of various familial diseases of muscle and brain and of inherited arrhythmias. The Na channelopathies were among the first molecularly characterized human ion channel diseases.11 Rare allelic variants in SCN5A have been linked to inherited ventricular arrhythmias,12 conduction system disease, and sudden infant death syndrome. Complex electrophysiological phenotypes have been associated with mutations in both α- and β-subunits of Na channels. Rare variants or mutations have been associated with sudden death in women (in a population-based study) and with atrial fibrillation. More common acquired forms of long QT syndrome (LQTS), generally associated with drug ingestion, have been linked to common variants of SCN5A. Finally, disease-causing mutations in the Na channel have been associated with alterations in drug blockade. The highly variant phenotypes of these arrhythmic syndromes are, in part, explained by the variable effects of the mutations on channel subunit function or expression.
Calcium Channels: L-type
The pore-forming subunit (α1) of the calcium (Ca) channel is built on the same structural framework as the Na channel.13 As is the case with the Na channel, a number of genes encode surface membrane Ca channel α1-subunits. The predominant sarcolemmal Ca channels in the heart are the L-type (large and long-lasting) and T-type (tiny and transient) Ca channels (Table 3-2). The cardiac L-type Ca channel (CaV1.2) is a multi-subunit transmembrane protein composed of α1C (α11.2) (165 kDa), β (55 kDa), and α2 (130 kDa) to δ (32 kDa) subunits. Three genes are known to encode L-type Ca channel α1-subunits (CaVα11.x to α13.x), and the CaVα11.2 is the gene expressed in the heart (see Table 3-1). Distinct splice variants of the CaVα11.2 gene have been described, and these contribute to the diversity of the cardiac L-type Ca channel function. Similar to the α-subunit of the Na channel, the S5-S6 linkers (P segments) of the α1-subunit of the Ca channel form the ion-selective pore (Figure 3-6). However, unlike Na channels, each P segment contributes a glutamic acid to a cluster that serve to bind Ca2+ in the channel pore. The β-subunit is completely cytoplasmic and noncovalently binds to the α1C subunit, modifying its function and contributing to appropriate membrane trafficking of the channel complex. Although β2a has been proposed to be the major L-type Ca channel β-subunit, splice variants of all β-subunits β1 to β4 are expressed in the mammalian heart in a complex spatial and temporal pattern. As many as five genes have been suggested to encode the α2-δ subunit: These gene products undergo post-translational processing to produce the mature extracellular α2-subunit linked by a disulfide bond to the transmembrane δ-subunit. In heterologous expression systems, α2– to δ-subunits enhance expression of Ca channels and hasten current activation and deactivation in the presence of α1– and β-subunits.
Table 3-2 Properties of Calcium Channels
| L-TYPE | T-TYPE | |
|---|---|---|
| Pore-forming α-subunit | α1C | α1H |
| Auxiliary subunits | β, α2-δ | ? |
| Permeability | Ba2+ > Ca2+ | Ba2+ ≅ Ca2+ |
| Activation threshold | >–30 mV | >–60 mV |
| Inactivation threshold | >–40 mV | >–90 mV |
| Inactivation | ||
| Rate | Slow | Fast |
| Calcium-dependent | Yes | No |
| Voltage-sensitive | Yes | Yes |
| Recovery | Fast | Slow |
| Localization in heart | All | Nodal > Purkinje > atria |
| Blocker sensitivity | ||
| Dihydropyridines | ++++ | + |
| Phenylalkylamines | ++++ | + |
| Benzothiazepines | ++++ | + |
| Tetralols | ++ | +++ |
| Ni2+ | + | +++ |
| Cd2+ | +++ | + |
Ba, Barium; Cd, cadmium; Ni, nickel.
Ca channels inactivate by both Ca2+-dependent inactivation (CDI) and voltage-dependent inactivation (VDI) processes. CDI and VDI are regulated by channel phosphorylation and β-subunits, which suggests shared structural mechanisms that perhaps involve the I-II domain linker. The C-terminus contains peptide sequences that bind calmodulin (CaM) and Ca2+; these are an IQ motif named for the signature amino acids (isoleucine and glutamine) in the sequence and a helix-loop-helix structural domain, referred to as an EF hand, that mediate CDI. CaM is permanently tethered to the channel complex and serves as a Ca2+ sensor for the L-type channel. The Ca2+-CaM complex facilitates the interaction of CaM with the IQ motif resulting in the occlusion of the inner mouth of the Ca channel pore and terminating the inward Ca2+ flux despite continued depolarization. As cytoplasmic Ca2+ concentration falls, calmodulin unbinds Ca2+ and the IQ motif, thus relieving Ca2+-dependent inactivation. Spatial discrimination of Ca2+ concentration by CaM may involve regions in the N-terminus of the channel. A Ca2+-binding EF hand motif in the carboxyl terminus of α1 appears to be necessary to confer Ca2+-dependent inactivation to the CaVα11.2-subunit, although not through a direct binding of Ca2+ (see Figure 3-6, bottom). The I-II interdomain linker has been demonstrated as the site of β-subunit binding as well as a critical structural determinant of VDI. Mutations in this linker have been associated with the highly arrhythmogenic Timothy syndrome.14
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


