Mitral Valve Regurgitation: Introduction
The normal mitral valve permits one-way blood flow from the left atrium to the left ventricle (LV) in an efficient, nearly frictionless fashion.1 Although even a normal competent valve may allow a trivial amount of reversed flow, more than a trace of mitral regurgitation is considered pathologic.2,3 Mild to moderate mitral regurgitation is tolerated indefinitely as long as it does not worsen.4 However, severe mitral regurgitation causes LV remodeling, reduced forward cardiac output, neurohumoral activation, LV damage, heart failure, and ultimately death.5 The natural history of mitral regurgitation depends intimately on its etiology, the severity of LV volume overload and its contractile performance, and the appearance of overlapping clinical conditions secondary to reversal of flow such as atrial fibrillation and pulmonary hypertension.6-8 In this setting, myxomatous degeneration of the mitral valve, a common pathologic substrate of mitral valve billowing (normal valve coaptation) and prolapse (deficient valve coaptation), is the most prevalent cause of isolated severe mitral regurgitation requiring surgical intervention in the United States.9 The following is a review of the normal mitral valve anatomy, as well as a summary of causes, consequences, and treatment of degenerative mitral valve regurgitation.
Mitral Valve Anatomy
The mitral valve is located in the left atrioventricular groove and allows unidirectional flow of oxygenated blood from the left atrium into the relaxed LV during diastole. The valve is a very complex three-dimensional assembly of separate anatomic components, including the annulus, the leaflets and commissures, the chordae, the papillary muscles, and the ventricle.10 During systolic contraction, a coordinated interaction of these anatomic components closes the valve against ventricular pressure. Therefore, its anatomy should be scrutinized systematically to identify the lesions (the abnormalities in valve structure) that lead to the valve’s dysfunction (the alteration in closure that results in mitral regurgitation).11
The mitral annulus is a fibromuscular ring located in the left atrioventricular groove that serves as an attachment and hinge point for the mitral valve leaflets. The mitral annulus is subjectively divided into anterior and posterior segments based on the attachments of the anterior and posterior mitral leaflets, but it can also be segmented by location into septal and lateral components. The anterior portion of the mitral annulus is in continuity with the fibrous skeleton of the heart, defined by the right and left fibrous trigones and the aortic mitral curtain. This portion of the mitral annulus is thus fibrous in nature and is much less prone to dilation in comparison to the posterior portion of the annulus (Fig. 77–1). Because the fibrous skeleton is discontinuous along the posterior portion of the mitral annulus, this portion dilates or increases its circumference in the setting of chronic mitral valve regurgitation, with associated atrial and ventricular dilatation.12 The resultant increase in mitral annular dimension makes the annulus more circular in shape, compared with its normal kidney bean shape, which in turn compromises the coaptation of the mitral leaflets due to the increase in septal-lateral or anterior-posterior dimension.13 The hinge point of the posterior portion of the mitral annulus may become atrialized in long-standing posterior leaflet prolapse and may also be affected by diffuse pathologic calcification.14 The normal mitral annulus also has a three-dimensional saddle shape, and the anterior portion of the annulus tends to bulge during systole to accommodate the aortic root. The overall circumference of the annulus may decrease by as much as 20% during systole, promoting central leaflet coaptation.15
Figure 77–1.
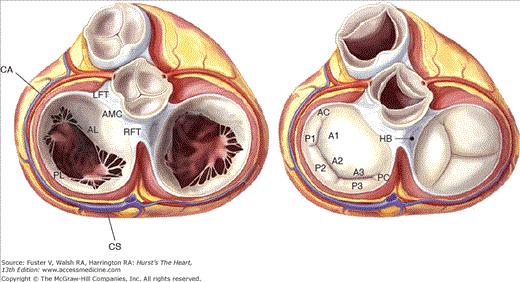
Anatomic view of the cardiac valves in diastole (left) and systole (right) with the left and right atrium cropped away and the great vessels transected. The illustration highlights the anatomic relations of the mitral valve, particularly its continuation with the aortic valve through the aorto-mitral curtain. AC, anterior commissure; AL, anterior leaflet; AMC, aorto-mitral curtain; CA, circumflex artery, CS, coronary sinus; HB, His bundle; LFT, left fibrous trigone (anterolateral trigone); PC, posterior commissure; PL, posterior leaflet; RFT, right fibrous trigone (posteromedial). Modified from Carpentierâs Reconstructive Valve Surgery by Carpentier AC, Adams DH, Filsoufi F (Saunders Elsevier, 2010).
The mitral valve has an anterior and posterior leaflet with similar surface areas but markedly different shapes.16 The anterior leaflet is taller than the posterior leaflet but with a shorter base, attaching to one-third of the annular circumference between the right and left fibrous trigones. During systole, the anterior leaflet forms a portion of the LV outflow tract through its continuity with the aorto-mitral curtain. The posterior leaflet is broader based, extending along the remaining two-thirds of the annulus, and has a shorter height. Despite their different shapes, the overall surface areas of the two leaflets are similar. The different orientations of the two leaflets ensure during systole that the closure line of the mitral valve will be located in the posterior one-third of the valve orifice, which prevents systolic anterior motion of the tip of the anterior leaflet into the outflow track.17 Both leaflets present two zones from their base to the free margin: the body zone (smooth and translucent) and the coaptation zone (thicker and rough due to the attachment of numerous chordae). During systole, the coaptation zones of the respective leaflets join together to form a seal from anywhere from a few millimeters to a centimeter, ensuring mitral valve competence (Fig. 77–2). The leaflets of the mitral valve can be “segmented” by location of the clefts or indentations in the posterior leaflet, which subdivide it into individual “scallops.” The middle scallop of the posterior leaflet is designated as P2, and the adjacent lateral and medial scallops are designated as P1 and P3, respectively (see Fig. 77–1). The anterior leaflet does not typically have natural indentations, but the corresponding areas of this leaflet are designated by opposition to the segments in the posterior leaflet as A1, A2, and A3.18
Figure 77–2.
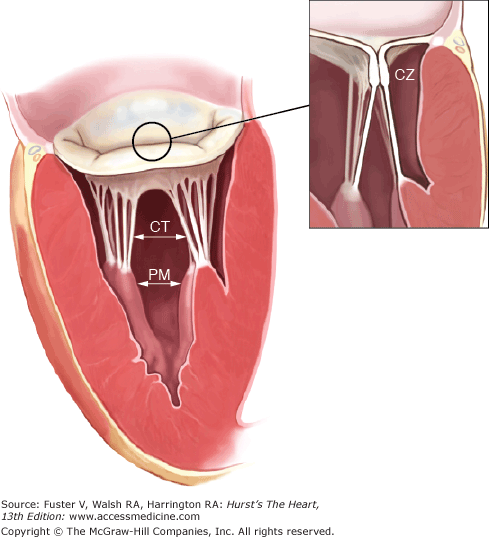
The mitral valve apparatus consists of the mitral leaflets, mitral annulus, chordae tendineae, the papillary muscles, and the left ventricle. Normal function of the mitral apparatus brings both leaflets together in systole and creates the coaptation zone. CT, chordae tendineae; CZ, coaptation zone; PM, papillary muscles. Modified from Carpentierâs Reconstructive Valve Surgery by Carpentier AC, Adams DH, Filsoufi F (Saunders Elsevier, 2010).
In addition to anterior and posterior leaflet segments, the mitral valve has posterior medial and anterior lateral commissures, which represent small segments of leaflet tissue presenting at the insertional junction of the anterior and posterior leaflets. These distinct areas of leaflet tissue are supported by chordal fans and are critical to ensure a good surface of coaptation at the junctions of the two leaflets. The height of commissural leaflet tissue can vary from a few millimeters to >1 cm.
The chordae tendineae attach the mitral leaflets to the papillary muscles and LV, creating a suspension system that allows full opening of the leaflets during diastole and prevents a displacement of the leaflets above the annular plane during systole. Chordae tendineae are classified according to their attachment between the free margin and the base of the leaflets.19 Primary or marginal chordae attach along the margin of the leaflets and are critical to prevent leaflet prolapse and to align the rough zone of the anterior and posterior leaflets during systole. Typically, primary chordae insert every 3 to 5 mm along the margin of both leaflets. Secondary, or body chordae attach to the ventricular side of the body of the leaflets and provide ventricular annular continuity as well as balancing of leaflet tension during systole. Tertiary, or basal chordae attach to the base of the leaflet hinge, providing additional linkage to the ventricle.20
The mitral valve leaflets are attached by the chordae tendineae to the papillary muscles, which are a part of the LV. The papillary muscles vary in the number of heads and exact position in the ventricle, but generally, there are two main groups comprising the anterior and posterior papillary muscles. Each papillary muscle is identified according to the relationship to the valve commissures, and each provides a fan chord to its corresponding commissure as well as to both anterior and posterior leaflets. The anterior papillary muscle’s blood supply can originate from both the left anterior descending artery and the circumflex artery, whereas the posterior papillary muscle is dependent primarily on the posterior descending artery. This explains the relative vulnerability of the posterior papillary muscle to ischemia and subsequent involvement in localized remodeling in the setting of ischemic mitral valve regurgitation. The LV supports the entire mitral apparatus due to the papillary muscles, and thus, ventricular dimensional changes in the setting of volume overload and remodeling, whether ischemic or not, can lead to leaflet tethering and mitral valve regurgitation.21 This important relationship of volume overload and remodeling to mitral valve dysfunction has led to the common observation that “mitral regurgitation begets mitral regurgitation.”
Degenerative Mitral Valve Regurgitation
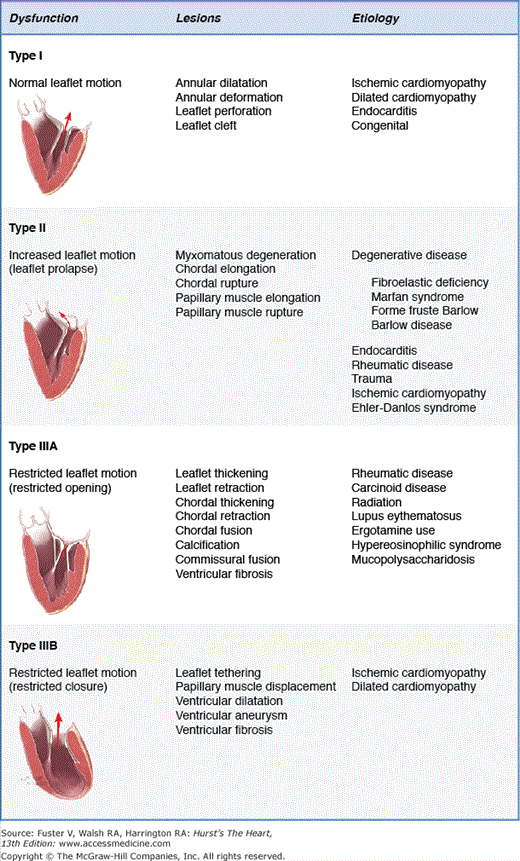
It is important to clarify the etiology and lesions that lead to clinically significant mitral valve regurgitation because treatment options and long-term outcomes vary in different clinical scenarios. It is also useful to identify the valve dysfunction that results from the lesions of the mitral valve apparatus. The main dysfunctions, lesions, and etiologies that can result in mitral valve regurgitation are shown in Fig. 77–3. Carpentier described this pathophysiologic triad, and it is a useful tool in everyday practice when assessing patients with mitral valve regurgitation.11,22 Dysfunctions are classified based on the position of the leaflet margins in relationship to the plane of the mitral annulus. Type I dysfunction implies normal leaflet motion, and the most common cause of significant mitral valve regurgitation is leaflet perforation or isolated annular dilatation, which is common in the setting of primary atrial fibrillation. Type II dysfunction implies excess leaflet motion and is most commonly associated with chordal elongation or rupture in the setting of degenerative mitral valve disease. Type IIIA dysfunction designates restricted opening and closing leaflet motion and results typically from rheumatic valve disease or other inflammatory scenarios that lead to chordal and leaflet scarring and calcification. Type IIIB dysfunction is associated with restricted leaflet motion in systole and is most commonly associated with papillary muscle displacement and associated leaflet tethering in the setting of ischemic or nonischemic dilated cardiomyopathy. Some others have chosen to designate conditions associated with types I, II, and IIIA dysfunction as primary or organic mitral regurgitation because the valve components (annulus, leaflets, and chords) are diseased, whereas type IIIB dysfunction is classified as secondary or functional mitral regurgitation because it is caused by perturbations in ventricular geometry.23
Although rheumatic heart disease is still the most common cause of mitral regurgitation worldwide, it is no longer a common cause of mitral regurgitation in developed countries.24 Additionally, although ischemic mitral regurgitation resulting from myocardial infarction still accounts for 10% to 20% of mitral regurgitation earlier intervention in acute coronary syndromes may be limiting the number of such cases in the future.25 degenerative mitral valve disease is now the leading cause of mitral valve disease and regurgitation.26 Degenerative mitral valve disease is defined by a spectrum of lesions, varying from simple chordal rupture involving prolapse of an isolated segment (particularly P2 or the middle scallop of the posterior leaflet) in an otherwise normal valve to multisegmental prolapse involving one or both leaflets in a valve with significant excess tissue and a large annular size (Fig. 77–4). Thus, the spectrum of degenerative disease is evident in clinical practice, which carries important surgical and clinical implications. Furthermore, based on this spectrum of lesions, degenerative disease may be further divided into two main entities—fibroelastic deficiency and Barlow disease27-29 (Fig. 77–5). Other terms used to describe degenerative mitral valve disease, including floppy valve syndrome, mitral valve prolapse, click-murmur syndrome, and parachute valve, cause much more confusion.30 For instance, mitral valve prolapse can cause a click and murmur on physical examination, but the terms fail to clarify etiology.31
Figure 77–4.
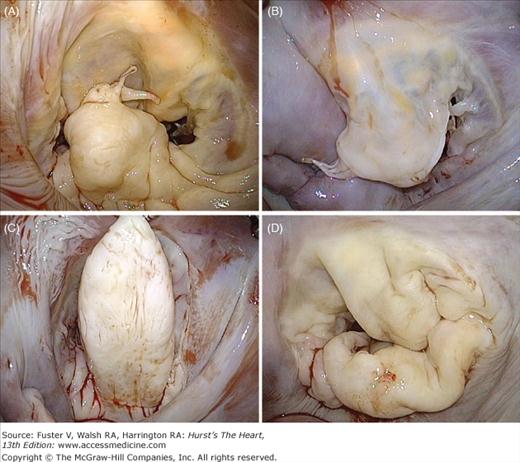
Valve lesions in degenerative mitral valve disease. A. Fibroelastic deficiency; isolated P2 prolapse secondary to chordal rupture and mild segmental thickening. B. Fibroelastic deficiency; anterior leaflet prolapse due to multiple ruptured chordae. C. Barlow disease; very tall and thickened P2 segment with otherwise normal P1 and P3 segments. D. Barlow disease; large valve with redundant, thick, bulky leaflets. Note the fissures blurring of the junction between atrium and leaflet.
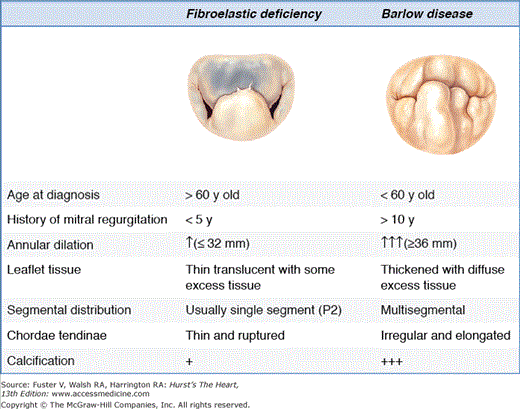
Fibroelastic deficiency generally occurs in patients over the age of 60 years.32 Generally, patients present with a relatively short history of valve disease, and mitral regurgitation is usually holosystolic and severe. Fibroelastic deficiency describes a condition associated with fibrillin deficiency that often leads to a rupture of one or more thinned and elongated chordae, usually involving the middle scallop of the posterior leaflet. Chordal rupture is the most common lesion causing mitral regurgitation in fibroelastic deficiency. Leaflets are usually thin and translucent, although the prolapsing segment may show myxomatous degeneration with leaflet segment thickening and distension in long-standing regurgitation. The key characteristic to make a distinction of fibroelastic deficiency within the spectrum of degenerative disease rests in the condition of the adjacent segments to the prolapsing segment, which are generally normal in size, height, and character.22,28,29 The valve annular size, as defined by anterior leaflet surface area, is generally ≥32 mm.
In contrast, patients with Barlow disease are generally younger (<60 years old) at the time of surgical referral and often present with a long history of follow-up for a murmur. Barlow valve disease causes a more diffuse and complex redundancy of the valve, producing prolapse and myxomatous degeneration of multiple segments in one or both leaflets (see Fig. 77–5). The most common lesions are excess leaflet tissue and leaflet thickening and distention, with diffuse chordal elongation, thickening, and/or rupture. Severe annular dilatation with giant valve size is evident (≤36 mm).33 Additionally, varying degrees of annular calcification are often observed, as are subvalvular fibrosis and calcification of the papillary muscles, in particular the anterior papillary muscle.34
Histologically, no specific cause of these abnormalities has been defined, although increased incidence has been associated with some genetic abnormalities (Fig. 77–6). However, no one genetic variation still explains the pathology seen.35-37 There are clear abnormalities in the extracellular matrix and leaflet, and chordal strength is below normal38-40 (Fig. 77–7). Furthermore, increased activation of matrix metalloproteases (MMP), which seem to play an important role in leaflet enlargement, has been also observed. In this regard, a murine model of overexpression of MMP2 produced a phenotype similar to human degenerative mitral valve disease.41 It is likely that genetic abnormalities render the valve susceptible to the degenerative process, and after mitral regurgitation develops, it places progressively more hemodynamic stress on the valve, perpetually worsening the disease. At present, no useful strategies have emerged for preventing or slowing the progression of degenerative mitral regurgitation.
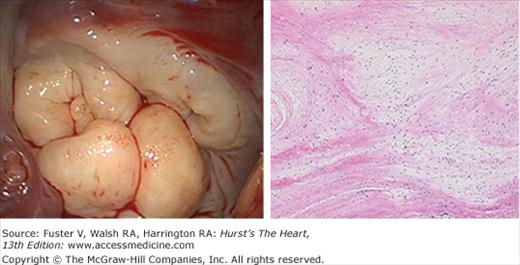
Figure 77–7.
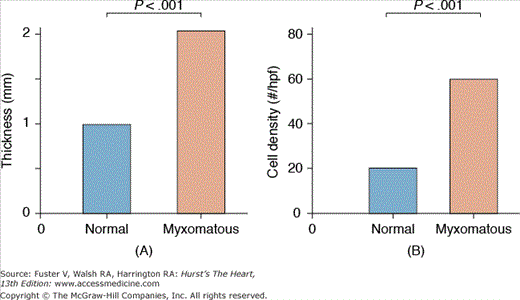
Quantitative analysis of mitral posterior leaflet tissue demonstrating significant thickening and increased cellularity of myxomatous valves. Hpf, high-power field. Data from Rabkin E, Aikawa M, Stone JR, et al. Activated interstitial myofibroblasts express catabolic enzymes and mediate matrix remodeling in myxomatous heart valves. Circulation. 2001;104:2525-2532.
Mitral regurgitation imparts a volume overload on the LV because it must compensate for the volume lost to regurgitation. Mild to moderate mitral regurgitation is well tolerated, possibly indefinitely, as long as the severity of mitral regurgitation does not increase. The grades of severity as suggested by the American College of Cardiology (ACC)/American Heart Association (AHA) Guidelines for the Management of Valvular Heart Disease42 are listed in (Table 77–1). Although these are only guidelines, they stem in part from the fact that when regurgitant fraction has been calculated for patients requiring mitral valve surgery, it almost always exceeds 50%.
| Mitral Valve Regurgitation | |||
|---|---|---|---|
| Mild | Moderate | Severe | |
| Qualitative | |||
| Angiographic grade | 1+ | 2+ | 3-4+ |
| Color Doppler jet area | Small central jet (< 4 cm2 or <20% LA size) | Signs of MR greater than mild but no severe MR | Vena contracta width >0.7 cm with large central jet (area >40% of LA) or with a wall-impinging jet swirling in LA |
| Doppler vena contracta width (cm) | <0.3 | 0.3-0.69 | ≥0.70 |
| Quantitative | |||
| Regurgitant volume (mL-beat) | <30 | 30-59 | ≥60 |
| Regurgitant fraction (%) | <30 | 30-49 | ≥50 |
| Regurgitant orifice area (cm2) | <0.20 | 0.20-0.39 | ≥0.40 |
| Additional criteria | |||
| Left atrial size | Enlarged | ||
| Left ventricular size | Enlarged | ||
Severe mitral regurgitation can be divided into three stages: acute, chronic compensated, and chronic decompensated43 (Fig. 77–8). In acute mitral regurgitation as might occur from rupture of marginal chordae tendineae, a small unprepared LV is suddenly confronted with a large volume overload from blood returning from the pulmonary veins summed with the regurgitant volume from the LV. The volume overload causes existing sarcomeres to stretch maximally, increasing end-diastolic volume and also stroke work through the Frank-Starling mechanism. The extra pathway for ejection into the left atrium unloads the LV, reducing end-systolic volume. Increased preload, decreased afterload, and a reflexive sympathetically mediated increase in contractility act in concert to increase total stroke volume and ejection fraction. However, because 50% or more of the total stroke volume is regurgitated into the left atrium, forward stroke volume and cardiac output are reduced. Additionally, the left atrium, which is of normal size and compliance, receives its very high total volume at high filling pressure, in turn leading to pulmonary congestion. Thus although LV function is normal or even supernormal, the patient suffers the low output and pulmonary congestion typical of LV failure. Many patients will require immediate corrective surgery at the time acute severe mitral regurgitation develops. In others, there may be a more gradual progression to severe mitral regurgitation so that it is better tolerated. Such patients may enter a chronic compensated phase. In this phase, eccentric hypertrophy has developed, increasing LV volume. Because the radius term in the Laplace equation for wall stress has increased (stress σ = p × r/2th, where p = LV pressure, r = radius, and th = thickness), afterload returns from subnormal to normal. However, increased preload and normal contractility permit a higher than normal ejection fraction of a large end-diastolic volume so that total stroke volume is greatly increased.44 This permits forward stroke volume to return to normal. Left atrial size is now enlarged, permitting it to accept the large regurgitant volume at nearly normal pressure. Thus the patient now has a near-normal cardiac output and filling pressure and is likely to be asymptomatic even during exercise. Although the patient may enjoy a period of compensation for years, eventually, contractile dysfunction sustained from prolonged hemodynamic overload ensues, and decompensation becomes manifest.45,46 Impaired contractility causes increased end-systolic volume and reduced stroke volume and cardiac output. Filling pressure is re-elevated, and the patient may develop heart failure symptoms. Although mitral regurgitation is usually thought to be a phenomenon that unloads the LV, in decompensated mitral regurgitation, the increased radius term in the Laplace equation causes systolic wall stress to increase, and afterload is greater than normal, contributing to LV dysfunction.47 In mitral regurgitation, increased LV radius is not offset by increased thickness, leading to the increase in wall stress. Thus, the relatively thin wall of the mitral regurgitant ventricle is beneficial to diastolic function and LV filling but is detrimental to LV systolic function because maladaptive LV remodeling causes increased afterload.48,49 It is important to note that ejection fraction may be held in the normal range by enhanced preload despite contractile dysfunction and afterload excess.
Figure 77–8.
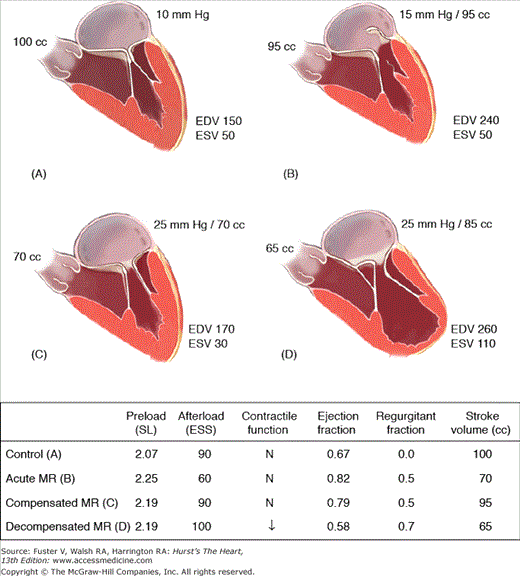
Normal physiology (control) is compared with that of acute mitral regurgitation (chordal rupture), compensated mitral regurgitation, and decompensated chronic mitral regurgitation starting with the upper left hand panel and going counterclockwise. The sudden opening of a new pathway for regurgitant flow into the left atrium increases left atrial pressure and preload (sarcomere length), in turn mildly increasing end-diastolic volume because resting sarcomere length is still 90% of maximum length. Afterload (end-systolic stress) is decreased, allowing more complete left ventricular ejection fraction and reducing end-systolic volume. These changes in loading increase ejection fraction and total stroke volume, but because 50% of the total stroke volume is lost to regurgitation (regurgitant fraction), forward stroke volume is decreased. Therefore, despite normal contractile fraction and increased ejection fraction, the patient presents with the hemodynamics of congestive heart failure. In chronic compensated mitral regurgitation (upper right), eccentric hypertrophy leads to substantial LV enlargement allowing it to pump extra volume, in turn resetting forward stroke volume toward normal. Enlargement of the left atrium allows it to accommodate the regurgitant volume at lower filling pressure. In the presence of decompensated chronic mitral regurgitation, muscle damage caused by prolonged severe volume overload reduces the effectiveness of ventricular ejection, and end-systolic volume increases. There is a further increase in diastolic volume, which is not compensatory, resulting in a decrease in total and forward stroke volumes. EDV, end-diastolic volume; ESS, end-systolic stress; ESV, end-systolic volume; MR, mitral regurgitation; SL, sarcomere length. Adapted from OâGara P, Sugeng L, Lang R, et al. The role of imaging in chronic degenerative mitral regurgitation. JACC Cardiovasc Imaging. 2008;1(2):221-237.
The LV dysfunction caused by severe mitral regurgitation stems from multiple pathologic processes. At the cellular level, there is loss of contractile elements in the endocardium in experimental models of mitral regurgitation and in the papillary muscles of humans.50,51 This abnormality can be reversed by valve repair/replacement in the experimental animal and in man and also by administration of β-blockers in the experimental animal.52-54 These data suggest that sympathetic overdrive, which is present in both human and experimental mitral regurgitation, contributes to the cellular pathology of the disease.55,56 In addition, the force-frequency relationship of the mitral regurgitant ventricle is depressed but can be normalized by the administration of forskolin, suggesting that abnormalities in calcium handling contribute to LV dysfunction.57
The LV remodeling of mitral regurgitation is unique and probably dictated by the loading conditions present. Mitral regurgitation stands out as a pure volume overload.58 In most other volume overloads such as anemia, heart block, and aortic regurgitation, the extra volume generated by the LV is ejected into the aorta, where the high stroke volume generates a widened pulse pressure and an element of systolic hypertension. Thus, most volume overloads are in fact a combination of volume and pressure overload, and the LV remodels accordingly. In aortic regurgitation, for instance, not only is LV volume increased to compensate for the regurgitated volume, but also LV thickness is greater than normal.59 Conversely, in mitral regurgitation, the extra volume is ejected into the left atrium, and systolic pressure is often low-normal. In turn, LV thickness is low-normal, producing a thin-walled, large LV, as noted earlier.
Several decades ago, Grossman et al60 proposed a paradigm for LV remodeling wherein the increased systolic wall stress of pressure overload was transduced to generate new sarcomeres laid down in parallel such that myocyte thickness and LV wall thickness increased. Increased wall thickness in the denominator of the Laplace equation offsets the increased pressure term in the numerator, keeping wall stress (afterload) normal and facilitating LV ejection. However, the increased diastolic stress from the sarcomere stretch of volume overload leads to new sarcomeres being laid down in series, increasing myocyte length and ventricular volume and allowing the ventricle to increase stroke volume. In experimental acute pressure overload, a 35% increase in contractile protein synthesis occurs within 6 hours of the onset of the pressure overload.61 Conversely, following the acute volume overload of mitral regurgitation and during chronic mitral regurgitation, no increase in protein synthesis has ever been detected.62 Because increased muscle mass can only accrue from either increased protein synthesis or decreased protein degradation and because synthesis is not increased, it has been suggested that the hypertrophy of mitral regurgitation develops from a process opposite of that of pressure overload (ie, decreased protein degradation instead of increased protein synthesis). It might be that older contractile proteins are less robust, a factor in part responsible for the LV dysfunction that ultimately develops.
As noted earlier, if mitral regurgitation is corrected before LV dysfunction is long standing, ventricular function can recover dramatically both in the experimental animal and in humans. Recovery is marked by restoration of myocyte contractile elements and a reduction in adrenergic drive.
In summary, the pure volume overload of mitral regurgitation is compensated by eccentric LV hypertrophy, which enables rapid LV diastolic filling and an increase in stroke volume. However, this remodeling eventually encumbers systolic emptying. This maladaptive geometry, together with the adrenergic overactivation, results in contractile protein loss, abnormal calcium handling, and a decrease in contractility. If mitral regurgitation is corrected in a timely fashion, this pathophysiology can be reversed.
The typical symptoms of mitral regurgitation are those of LV failure and include dyspnea on exertion, orthopnea, and paroxysmal nocturnal dyspnea. If pulmonary hypertension has developed, ascites and edema may also occur. Debate continues as to whether or not there is a mitral valve prolapse syndrome (ie, a group of symptoms associated with degenerative mitral valve disease). These symptoms are thought to include palpitation, fatigue, and chest pain, which are atypical of classic angina and syncope or presyncope.63,64 These symptoms are common in the general population, and whether they occur more frequently in patients with mitral valve prolapse continues to be a subject of controversy.
On physical examination, the reduced forward stroke volume tends to reduce systolic blood pressure and pulse pressure, but this finding is quite variable, and some patients are actually hypertensive. The apical beat is displaced downward and to the left in chronic severe disease due to LV dilatation. The typical murmur is holosystolic if the lesion is chordal rupture and is heard best at the apex and radiates to the axilla. There is a weak positive correlation between mitral regurgitation severity and murmur intensity.65 Severe mitral regurgitation is often accompanied by an S3 produced by the emptying of the large left atrial volume under higher than normal pressure into the LV. The presence of an S3 is often evidence that the mitral regurgitation is severe, rather than indicating that the patient is in heart failure.
Mitral valve prolapse in Barlow disease is sometimes referred to as click-murmur syndrome, indicative of the typical findings on physical examination of a mid-systolic click followed by a late systolic murmur. The click is generated as the elongated chordae are stretched taut. The valve leaflets then move past their coaptation point, and the murmur ensues. Physical maneuvers that decrease LV volume, such as standing or the Valsalva maneuver, cause the click and murmur to come earlier in systole and consequently to increase in intensity (Table 77–2). This occurs because a decrease in LV volume reduces tension on the mitral valve, in effect lengthening the valve apparatus. Maneuvers that increase LV volume, such as squatting or lying down, may cause the opposite effect or may cause the click and murmur to disappear all together. In some patients, only the click or murmur is present, or mitral valve prolapse may occur without any physical findings. As the severity of mitral regurgitation worsens, the murmur becomes progressively more holosystolic, and the click may disappear.
| Position | Timing | Intensity |
|---|---|---|
| Standing | ← | ↑ |
| Recumbent | → | ↓ or more |
| Squatting | → | ↓ or more |
| Hand grip | ← | ± |
| Valsalva | ← | ± |
| Amyl nitrite | ± | ↑ |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


