Metabolic Syndrome, Obesity, and Diet: Introduction
The rise in the prevalence of obesity in the United States and worldwide is threatening to undo recent advances in prevention of atherosclerotic cardiovascular disease (ASCVD). Between 1993 and 2008, the proportion of obese adults in the United States increased from 14.5% to 26.7%.1 Among the complications associated with obesity, cardiovascular events produce the greatest morbidity and mortality. A significant portion of the latter occurs in persons in whom obesity precedes type 2 diabetes. But diabetes is only one of several conditions that associate strongly with obesity. Others include dyslipidemia, hypertension, systemic inflammation, and a thrombotic tendency. Recently there has been a trend in the cardiovascular field to group all of these factors together under the heading of metabolic syndrome.2 In this sense, metabolic syndrome can be taken to represent a multiplex cardiovascular risk factor. This syndrome does not include, but is strongly associated with, other complications of obesity; for example, fatty liver, cholesterol gallstones, obstructive sleep apnea, and polycystic ovarian syndrome. The current definition generally regards hyperglycemia in the range of type 2 diabetes to be one of the components of metabolic syndrome.2,3 This is because approximately 85% of persons classified as having type 2 diabetes will meet current criteria for metabolic syndrome. Even so, many investigators in the diabetes field prefer to separate metabolic syndrome from diabetes and to view it largely as a prediabetic condition, besides being a cardiovascular risk factor.4,5 This is a semantic argument. Regardless of viewpoint, type 2 diabetes must be viewed as one of the complications of obesity and strongly associated with risk for ASCVD. This chapter focuses primarily on metabolic syndrome as a cardiovascular risk factor, with obesity being the primary exogenous factor driving its development. But other exogenous factors are further discussed, such as physical inactivity and dietary excesses, as well as endogenous susceptibility factors.
Metabolic syndrome represents a clustering of cardiovascular risk factors that are amalgamated into a single multiplex risk factor for ASCVD (Fig. 92–1).6 When the concept of metabolic syndrome was introduced into the National Cholesterol Education Program Adult Treatment Panel III Report (ATP III),2 it was considered a partner of elevated low-density lipoprotein (LDL) in the causation of cardiovascular disease (CVD). The metabolic risk factors that make up the syndrome include atherogenic dyslipidemia, elevated blood pressure, dysglycemia, a prothrombotic state, and a proinflammatory state. Several reports indicate that this clustering of metabolic risk factors cannot be explained by chance alone; hence the use of the term syndrome.7-20 This suggests that there is a common, underlying etiology of this clustering. Individuals with metabolic syndrome have an approximate doubling of risk for ASCVD and approximately four-fold higher risk for developing type 2 diabetes compared with those without the syndrome.
Figure 92–1.
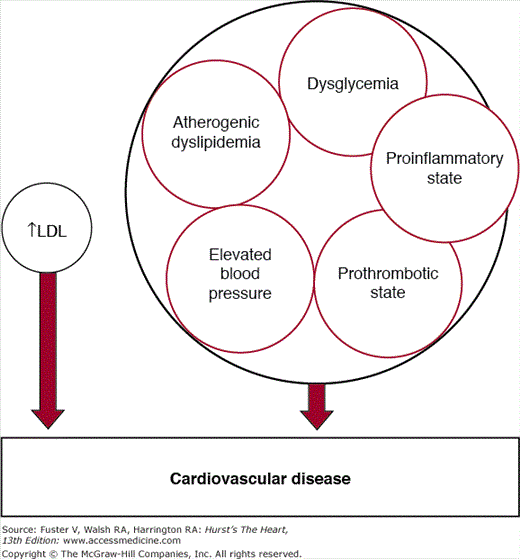
Risk factor partners: elevated low-density lipoprotein (LDL) and metabolic syndrome. The latter is a multiplex risk factor for arteriosclerotic cardiovascular disease and a clustering of atherogenic dyslipidemia, elevated blood pressure, dysglycemia, a prothrombotic state, and a proinflammatory state.
Low-Density Lipoprotein and Metabolic Syndrome: Partners in Atherogenesis
The development of atherosclerosis can be considered to occur in 2 stages: injury and response to injury. The primary injurious agents include LDL and other apolipoprotein B (apo B)–containing lipoproteins. The response to injury makes up a process called inflammation. Metabolic syndrome exacerbates atherogenesis by enhancing the inflammatory response to LDL injury. The key steps in both processes are reviewed briefly.
The first step in the pathogenesis of atherosclerosis is the infiltration of plasma LDL into the arterial intima (Fig. 92–2). The rate of infiltration of LDL depends on 2 factors: (1) the concentration of LDL in the circulation, and (2) the permeability of the arterial wall.21 Several mechanisms have been proposed for transport into the subendothelium: vesicular ferrying through endothelial cells, passive sieving through endothelial-cell pores, and passage between cells. Not all that enters the arterial wall stays there. Some escapes by a reversal of the same process. However, a portion of the LDL becomes entrapped into the extracellular matrix.22 When this occurs, LDL is ripe for modification. Several types of modification have been proposed: aggregation, fusion of lipoproteins, proteolysis, lipolytic degradation such as hydrolysis of cholesterol esters, phospholipids, and triglyceride, oxidation and glycation.23 When LDL is modified in various ways, it acquires inflammatory potential. The consequences of LDL modification include the activation of various types of cells—endothelial cells, monocyte/macrophages, and smooth muscle cells.22,23 All of these changes come under the category of inflammation (Fig. 92–3). Key changes are endothelial dysfunction, which allows for a more rapid infiltration of LDL into the arterial wall and adherence to circulating monocytes; movement of monocytes into the arterial wall and their activation; proliferation of smooth muscle cells; and enhanced fibrosis. Macrophages are a key player in atherogenesis.24 They first accumulate lipid and then undergo apoptosis—releasing their excess lipid into lipid pools. Macrophages further produce enzymes, such as metalloproteinases, that degrade the extracellular matrix. These latter two changes seemingly create unstable plaques that are prone to rupture and to causation of acute ASCVD events.
Figure 92–2.
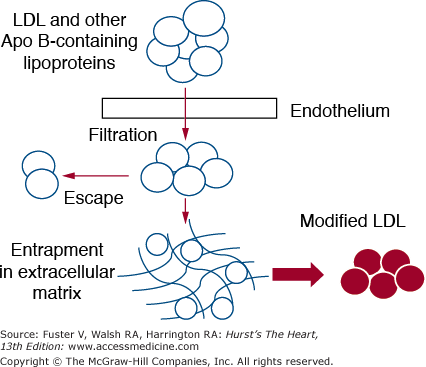
Details of arterial injury produced by low-density lipoprotein (LDL) and other apolipoprotein B (apo B)–containing lipoproteins. Circulating lipoproteins filter into the arterial intima through the endothelium. A portion of LDL escapes back into the circulation. Other LDL particles, however, become entrapped in extracellular matrix. Here they become modified in several ways; this modification occurs in ways that turn the LDL into proinflammatory agents.
Figure 92–3.

Details of the inflammatory process evoked by modified low-density lipoprotein (LDL). Three cellular systems are affected by modified LDL: endothelial cells, monocytes/macrophages, and smooth muscle cells. Modified LDL causes endothelial dysfunction, allowing increased amounts of LDL to filter into the arterial wall and enhanced attachment of monocytes to the endothelium. It also acts as a chemoattractant to pull monocytes into the arterial wall; at the same time it promotes transformation of monocytes into macrophages and activates them. Activated macrophages ingest modified LDL, become transformed into foam cells, undergo apoptosis to form large lipid pools, and release metalloproteinases to degrade the extracellular matrix. The latter two effects lead to destabilization of arteriosclerotic plaques, plaque rupture, and acute cardiovascular events. Finally, modified LDL stimulate smooth muscle cell proliferation for production of collagen fibers, leading to fibrosis of the plaque.
Metabolic Syndrome and Arterial Inflammation
Whereas excess LDL initiates atherogenesis and promotes its progression, metabolic syndrome exacerbates the inflammatory process. This leads to a worsening acceleration of atherosclerosis. Each of the components of metabolic syndrome appears to worsen inflammation in plaques. First, in the case of hypertension, an increased hydrostatic pressure in elevated blood pressure can enhance influx of LDL into the arterial wall. Further, hypertension is associated with endothelial dysfunction.25 Hypertension can be accompanied by increased angiotensin II (A-II), which can enhance leukocytal vascular adhesion molecule (VCAM)-1 on endothelial cells and cause release of proinflammatory cytokines (eg, interleukin-6 [IL-6] and monocyte chemotactic protein-1 [MCP-1]).26,27 A-II can also increase the expression by arterial smooth muscle cells of proinflammatory cytokines such as IL-6 and MCP-1 and of the leukocyte adhesion molecule VCAM-1 on endothelial cells.26,27 Second, dysglycemia, particularly diabetic hyperglycemia, has been implicated in several ways in the exacerbation of inflammation—formation of inflammatory advanced glycation products, glycation of extracellular matrix enhancing retention of LDL, glycoxidative modification of LDL, and activation of protein kinase C (enhancing the inflammatory response).28 Third, a key component of atherogenic dyslipidemia is a low high-density lipoprotein (HDL). HDL is believed to be a protective lipoprotein, and if so, most likely exerts its antiinflammatory effects at multiple levels: It transports excess cholesterol out of macrophages, reducing their atherogenic potential; it prevents conversion of LDL into proinflammatory modified LDL; and it inhibits cytokine-induced expression of cellular adhesion molecules on endothelial cells.29 At present, the role of HDL in the inflammatory process is still under intense investigation.30,31 The protective actions of HDL are reduced in persons with atherogenic dyslipidemia. Fourth, in metabolic syndrome, there is an increase in circulating cytokines.32,33 These cytokines likely act at the level of the arterial wall to enhance the inflammatory response to modified LDL. And finally, a prothrombotic state is characterized by a series of abnormalities that can enhance coagulation, inhibit fibrinolysis, and alter platelet function. Among these factors are increases in plasminogen activator inhibitor-1, fibrinogen, factor VII, factor VIII, factor X, prothrombin fragments F1+2, and von Willebrand factor.34,35 These factors can not only promote inflammation within arteriosclerotic plaques, but they can enhance thrombus propagation after a ruptured plaque.
Pathogenesis of Metabolic Syndrome
A simple way to visualize the pathogenesis of metabolic syndrome is illustrated in Fig. 92–4. This view identifies an interaction between exogenous and endogenous factors. Obesity is the major exogenous factor, but physical inactivity and excess dietary factors can play a role. Endogenous factors include inherent insulin resistance, dysfunctional adipose tissue, endocrine disorders, and various genetic aberrations. The endogenous factors can be grouped together under the heading of metabolic susceptibility. To develop the syndrome, most individuals must be metabolically susceptible. But even in the presence of susceptibility, the full-blown metabolic syndrome generally will not develop in the absence of exogenous factors (especially obesity).
Figure 92–4.

Pathogenic scheme for development of the metabolic syndrome. The syndrome develops as a result of the interaction of exogenous and endogenous factors. The major exogenous factor is obesity, but physical inactivity and atherogenic diet play an important role. Endogenous factors include dysfunctional adipose tissue, genetic forms of insulin resistance, various endocrine disorders, and other genetic susceptibility.
The high prevalence of metabolic syndrome in the United States36 and worldwide37 is secondary to a rising prevalence of obesity.38-42 Metabolic syndrome prevalence rises in parallel with increasing obesity.43 Physical inactivity also is associated with a higher prevalence of metabolic syndrome.44-51 Part of this association can be related to the greater obesity accompanying a sedentary lifestyle; nevertheless, it is likely that physical activity provides a protective role against metabolic syndrome independently of the obesity. Further, high-carbohydrate diets, particularly those rich in simple carbohydrates or high-glycemic index foods, have been claimed to worsen metabolic syndrome.52-55 The literature nonetheless is mixed on the ideal diet composition for prevention and treatment of the syndrome.52
The mechanisms whereby obesity results in metabolic syndrome are being increasingly understood.2 Adipose tissue releases several products that appear to worsen metabolic syndrome.56 The most important is a key fuel source, nonesterified fatty acids (NEFA). During the fasting state, adipose tissue triglyceride undergoes lipolysis and releases NEFA into the circulation. The major enzyme involved in lipolysis is hormone-sensitive lipase; the activity of this enzyme is enhanced by catecholamines and suppressed by insulin. When insulin levels are low during fasting, lipolysis is high, as is NEFA release. NEFA is the major energy source during fasting. But if NEFA supply exceeds needs for energy utilization, lipid accumulates in muscle and liver. This accumulation is called ectopic fat. When fat accumulates in muscle and the liver, insulin resistance is increased. This change plus other metabolic alterations predisposes to the metabolic syndrome.56
Beyond excess fatty acids, other products of adipose tissue are released in abnormal amounts from adipose tissue. One category of products includes the inflammatory cytokines; for example, tumor necrosis factor-α (TNF-α) and IL-6.57 Excess cytokine release appears to be secondary to infiltration of adipose tissue with activated macrophages, which can produce these cytokines.58 The result is a high level of circulating cytokines. These can have several systemic effects: enhancement of insulin resistance in muscle, production of acute-phase reactants (C-reactive protein [CRP] and fibrinogen) by the liver, and exacerbation of inflammation in arteriosclerotic lesions. Both of the latter can predispose to major cardiovascular events. These cytokines play a key role in the causation of the proinflammatory state of metabolic syndrome.
The adipose tissue likewise can predispose to a prothrombotic state by release of excess amounts of plasminogen activator inhibitor-1, which is released from adipose tissue in response to obesity.59 Adipose tissue further secretes leptin, an appetite suppressant. Leptin levels are high in obesity and seemingly do not suppress the appetite of obese individuals, a condition called leptin resistance. Leptin can have systemic actions as well as actions in the hypothalamus. One such systemic action is to enhance fatty-acid oxidation by the liver, preventing steatosis.60 Several other bioactive adipokines have been reported to be produced by adipose tissue: resistin, angiotensinogen, tissue factor, transforming growth factor β, nitric oxide synthase, acylation stimulating protein, adipophilin, adipoQ, adipsin, monobutyrin, and agouti protein. Their role in the causation of metabolic syndrome remains to be fully elucidated.
In adipose tissue, 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) converts inactive cortisone to active cortisol. Overexpression of 11β-HSD1 induced in mice produces central obesity and insulin resistance.61 It also has been reported that obesity in humans is accompanied by overexpression of 11β-HSD1.62
Finally, the release of another substance, adiponectin, is actually reduced with obesity.63-66 Adiponectin can protect against insulin resistance, metabolic risk factors, and atherogenesis. The mechanisms whereby adiponectin exerts this protective effect is a topic of intense research at present.
Only a portion of patients with obesity develop metabolic syndrome. It appears that an individual must be metabolically susceptible to developing the syndrome, and when obesity is acquired, the syndrome becomes manifest. Several factors seemingly contribute to endogenous susceptibility. Among these are dysfunctional adipose tissue, genetic forms of insulin resistance, various endocrine disorders, and other genetic factors. Of particular importance appears to be a dysfunction of adipose tissue.
There are at least four potential disorders that can contribute to dysfunctional adipose tissue, which in turn will accentuate metabolic syndrome. These include a deficiency of subcutaneous adipose tissue, genetic forms of insulin resistance, dysfunctional adipocytes, and inflammation of adipose tissue (Fig. 92–5).
Figure 92–5.
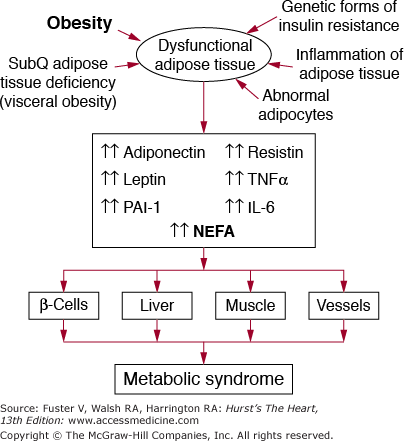
The role of obesity and dysfunctional adipose tissue in causation of the metabolic syndrome. When adipose tissue becomes overloaded with lipid (obesity), it produces abnormal amounts of nonesterified fatty acids (NEFA) and other adipokines. Among the latter are adiponectin, leptin, plasminogen activator inhibitor-1 (PAI-1), resistin, tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), and other inflammatory cytokines. The protective adiponectin is produced in subnormal amounts in obese persons. These abnormal products of adipose tissue flood various key tissues—pancreatic β cells, liver, muscle, and vessels; their effects in turn give rise to metabolic syndrome or accelerate the development of atherosclerosis at the level of the arterial wall. Additional causes of dysfunctional adipose tissue include a deficiency of subcutaneous adipose tissue, leading to visceral obesity, genetic forms of insulin resistance, abnormal adipocytes, and inflammation of adipose tissue. In the presence of these factors, defects in production of NEFA and adipokines is accentuated, worsening the metabolic syndrome.
One of the most important of these is a deficiency of subcutaneous adipose tissue. This abnormality is seen in an extreme form in a condition called lipodystrophy. Several metabolic defects can cause lipodystrophy—a severe deficiency of adipose tissue.67 In patients with lipodystrophy, who have little adipose tissue for storage of extra energy, fat becomes deposited ectopically in the liver and muscle; the result is the development of severe metabolic syndrome. Less severe forms of adipose-tissue deficiency are manifest by an abnormal body fat distribution. Differences in body fat distribution can typically be seen between obese women and men.68 Women normally have considerable quantities of subcutaneous adipose tissue in the lower body. Only when they are more severely obese does fat begin to accumulate in the upper body. It first enters upper body subcutaneous adipose tissue and only later does it accumulate in visceral adipose tissue beds. As a result, substantial ectopic fat accumulation is relatively rare. In contrast, men typically have a paucity of lower-body subcutaneous adipose tissue; as a result, they tend to develop upper-body obesity, including considerable amounts of visceral fat as well as ectopic fat. This pattern of fat distribution is called abdominal obesity; it is complicated by larger amounts of ectopic fat, which predisposes to metabolic syndrome.69 There is considerable variation in these trends in both men and women, and some individuals are particularly prone to development of ectopic fat and metabolic syndrome when they become obese.68 The causes of a relative deficiency of subcutaneous adipose tissue are not known, although because of male/female differences, endocrine factors can contribute. The net result of adipose tissue deficiency is a shift of fat away from adipose tissue and into ectopic stores, which will worsen metabolic syndrome. In addition, the normal release of other adipokines appears to be impaired.70
Dysfunctional forms of adipose tissue further can result from genetic forms of insulin resistance. Insulin is a major regulator of adipose tissue metabolism. When genetic defects occur in insulin signaling in adipocytes, suppression of lipolysis and other products is impaired.71 In addition, adiponectin release is reduced.71 All of these will accentuate ectopic fat distribution and metabolic syndrome. Moreover, defective insulin signaling in other tissues such as muscle and liver most likely will accentuate metabolic syndrome.72,73 A good example of a genetic form of insulin resistance is found in many persons of South Asian origin. Insulin-resistant South Asians have multiple signs of dysfunctional adipose tissue—elevated NEFA levels, high CRP and leptin levels, and low adiponectin concentrations—even when they are not obese by European or North American standards.71 These persons are prone to metabolic syndrome and to premature type 2 diabetes and CVD.
It is likely that dysfunction within adipocytes contributes to failure to store fat, to suppress lipolysis, or to suppress release of other adipokines. Defects in adipocyte function might occur at several levels, including conversion of mesenchymal stem cells into preadipocyte, further conversion into various adipocyte populations, and adipocyte cell death.74 A variety of key pathways have been described in adipocytes in which defects potentially could lead to abnormalities in product release.75-80
Finally, in obese persons, the adipose tissue is invaded with macrophages.58,81-84 The possibility has been raised that activation of these macrophages will result in the production of cytokines that will derange the function of adipocytes. In particular, these cytokines can cause insulin resistance, and the same defects are noted in persons with genetic forms of insulin resistance. Thus inflammation of adipose tissue can be yet another factor contributing to dysfunctional adipose tissue and metabolic syndrome.
In the preceding discussion, the effects of genetic forms of insulin resistance on adipose tissue were reviewed. One hypothesis holds that genetic forms of insulin resistance are the major cause of metabolic syndrome.85,86 According to this hypothesis, resistance to the action of insulin is widespread and causes a gross metabolic disturbance in many tissues. This disturbance can account for the multiple metabolic risk factors characteristic of the syndrome. This hypothesis is provocative and has provided a basis for many studies on the causation of metabolic syndrome. The effects of insulin resistance in adipose tissue provide the most direct evidence for the mechanism linking resistance to insulin to metabolic syndrome. Nevertheless, it is certainly possible that widespread metabolic disturbance contributes beyond adipose tissue abnormalities.85 Just how much of metabolic syndrome can be attributed to genetic forms of insulin resistance is uncertain. However, the close association between obesity and dysfunctional adipose tissue and the syndrome suggests that in the overall picture, genetic forms of insulin resistance are not dominant. Nonetheless, insulin resistance can be a particularly important contributor to the syndrome if it is present in conjunction with obesity.86
In view of metabolic differences in men and women, it is likely that endocrine factors play a role in causation of metabolic syndrome. This possibility is heightened by the observation that women with polycystic ovary syndrome are prone to metabolic syndrome.87-89 Because patients with hypercorticoidism manifest many of the features of the syndrome, abnormalities in cortisol metabolism also have been implicated.90
Manifestations of metabolic syndrome vary from individual to individual and also between populations. For example, Asians and Hispanics appear to be particularly susceptible to diabetes, African Americans to hypertension, and Caucasians to dyslipidemia. Certainly all of the features of metabolic syndrome can occur in all of these populations, but prominent features suggest that genetic variation exists and affects manifestations of the syndrome. Research on the genetic basis of ethnic differences has been increasing the past few years and promises to provide new insights into the causes of variation in expression of the syndrome.
In 2005, the American Heart Association (AHA) and the National Heart, Lung, and Blood Institute (NHLBI) updated the ATP III criteria.2 The essential criteria of ATP III were retained. Of note, however, they lowered the threshold for impaired fasting glucose to 100 mg/dL from the previous 110 mg/dL, in accord with current American Diabetes Association recommendations.91 The International Diabetes Federation92,93 in 2005 published similar criteria for the diagnosis of metabolic syndrome. In 2009 the AHA, NHLBI, and International Diabetes Federation, along with the World Heart Federation, International Atherosclerosis Society, and the International Association for the Study of Obesity, issued a joint interim statement regarding a common universal definition for the metabolic syndrome.3 They agreed that the requirement for central obesity as an obligatory component should be dropped, but that waist circumference would continue to be a useful preliminary screening tool. Three of 5 abnormal findings are required for the diagnosis of the metabolic syndrome. With the exception of waist circumference, where national or regional cut points may be used pending further research, a single set of cut points should be used for all other components of the metabolic syndrome (Table 92–1). The current recommended waist circumference thresholds for abdominal obesity vary by region and ethnicity (Table 92–2).
| Measure | Categorical Cut Points |
|---|---|
| Elevated waist circumferencea | Population- and country-specific definitions |
| Elevated triglycerides (drug treatment for elevated triglycerides is an alternate indicatorb) | ≥150 mg/dL (1.7 mmol/L) |
| Reduced HDL-C (drug treatment for reduced HDL-C is an alternate indicatorb) | <40 mg/dL (1.0 mmol/L) in males; <50 mg/dL (1.3 mmol/L) in females |
| Elevated blood pressure (antihypertensive drug treatment in a patient with a history of hypertension is an alternate indicator) | Systolic ≥130 and/or diastolic ≥85 mm Hg |
| Elevated fasting glucosec (drug treatment of elevated glucose is an alternate indicator) | ≥100 mg/dL |
| Recommended Waist Circumference Threshold for Abdominal Obesity | ||||
|---|---|---|---|---|
| Population | Organization (Reference) | Men | Women | |
| Europid | IDF (4) | ≥94 cm | ≥80 cm | |
| Caucasian | WHO (7) | ≥94 cm (increased risk) ≥102 cm (still higher risk) | ≥80 cm (increased risk) ≥88 cm (still higher risk) | |
| United States | AHA/NHLBI (ATP IIIa (5) | ≥102 cm | ≥88 cm | |
| Canada | Health Canada (8,9) | ≥102 cm | ≥88 cm | |
| European | European Cardiovascular Societies (10) | ≥102 cm | ≥88 cm | |
| Asian (including Japanese) | IDF (4) | ≥90 cm | ≥80 cm | |
| Asian | WHO (11) | ≥90 cm | ≥80 cm | |
| Japanese | Japanese Obesity Society (12) | ≥85 cm | ≥90 cm | |
| China | Cooperative Task Force (13) | ≥85 cm | ≥80 cm | |
| Middle East, Mediterranean | IDF (4) | ≥94 cm | ≥80 cm | |
| Sub-Saharan African | IDF (4) | ≥94 cm | ≥80 cm | |
| Ethnic Central and South American | IDF (4) | ≥90 cm | ≥80 cm | |
In populations at risk, metabolic syndrome is accompanied by an increase in relative risk for ASCVD.94-103 In prospective epidemiologic studies, the relative risk for ASCVD events is essentially doubled. It is likely that the two-fold increase in risk seen in short-term, prospective studies underestimates the long-term impact of the syndrome. The reason is that metabolic risk factors tend to worsen with time. Lipid levels and blood pressure rise with advancing age, and normal glucose levels advance to prediabetes or frank diabetes. Consequently, the earlier metabolic syndrome can be detected and managed, the slower will be the progression.
At present, more intense clinical intervention is driven by short-term risk for ASCVD.6 This risk usually is identified as 10-year risk for coronary heart disease (CHD). According to ATP III guidelines, risk can be stratified into 4 categories:
High risk is a 10-year risk for CHD >20% and includes patients with clinically evident ASCVD, diabetes, or enough other major risk factors to raise the risk to this level.
Moderately high risk consists of ≥2 major risk factors and a 10-year risk of 10% to 20%.
Moderate risk exhibits ≥2 risk factors, but a 10-year risk <10%.
Lower-risk individuals have 0 to 1 risk factor and a 10-year risk <10%.
Most persons with metabolic syndrome can be considered to be at least a moderate risk; but many will have risk >10%.
Framingham risk scoring should be used to estimate 10-year risk in metabolic syndrome patients without established ASCVD or type 2 diabetes mellitus.6 Because metabolic syndrome is only one part of overall risk assessment for ASCVD, it is not an adequate tool to estimate 10-year risk for CHD. These patients must be considered to be at higher lifetime risk for ASCVD, but metabolic syndrome alone is inadequate to guide clinical management for short-term risk reduction.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


