Severity
Clinical circumstances
A. Develops in presence of extracardiac condition that intensifies myocardial ischemia (secondary UA)
B. Develops in absence of extracardiac condition (primary UA)
C. Develops within 2 week after AMI (postinfarction UA)
I. New onset of severe angina or accelerated angina; no rest pain
IA
IB
IC
II. Angina at rest within past month but not within preceding 48 h (angina at rest, subacute)
IIA
IIB
IIC
III. Angina at rest within 48 h (angina at rest, acute)
IIIA
IIIB
IIIC
Table 18.2
Definition of myocardial infarction
Criteria for acute myocardial infarction |
The term acute myocardial infarction (MI) should be used when there is evidence of myocardial necrosis in a clinical setting consistent with acute myocardial ischemia. Under these conditions any one of the following criteria meets the diagnosis for MI: |
Detection of a rise and/or fall of cardiac biomarker values [preferably cardiac troponin (cTn)] with at least one value above the 99th percentile upper reference limit (URL) and with at least one of the following: |
Symptoms of ischemia. |
New or presumed new significant ST-segment–T wave (ST–T) changes or new left bundle branch block (LBBB). |
Development of pathological Q waves in the ECG. |
Imaging evidence of new loss of viable myocardium or new regional wall motion abnormality. |
Identification of an intracoronary thrombus by angiography or autopsy. |
Cardiac death with symptoms suggestive of myocardial ischemia and presumed new ischemic ECG changes or new LBBB, but death occurred before cardiac biomarkers were obtained, or before cardiac biomarker values would be increased. |
Percutaneous coronary intervention (PCI) related MI is arbitrarily defined by elevation of cTn values (>5 × 99th percentile URL) in patients with normal baseline values (≤99th percentile URL) or a rise of cTn values >20 % if the baseline values are elevated and are stable or falling. In addition, either (i) symptoms suggestive of myocardial ischemia or (ii) new ischemic ECG changes or (iii) angiographic findings consistent with a procedural complication or (iv) imaging demonstration of new loss of viable myocardium or new regional wall motion abnormality are required. |
Stent thrombosis associated with MI when detected by coronary angiography or autopsy in the setting of myocardial ischemia and with a rise and/or fall of cardiac biomarker values with at least one value above the 99th percentile URL. |
Coronary artery bypass grafting (CABG) related MI is arbitrarily defined by elevation of cardiac biomarker values (>10 × 99th percentile URL) in patients with normal baseline cTn values (≤99th percentile URL). In addition, either (i) new pathological Q waves or new LBBB, or (ii) angiographically documented new graft or new native coronary artery occlusion, or (iii) imaging evidence of new loss of viable myocardium or new regional wall motion abnormality. |
Criteria for prior myocardial infarction |
Any one of the following criteria meets the diagnosis for prior MI: |
Pathological Q waves with or without symptoms in the absence of non-ischemic causes. |
Imaging evidence of a region of loss of viable myocardium that is thinned and fails to contract, in the absence of a non-ischemic cause. |
Pathological findings of a prior MI. |
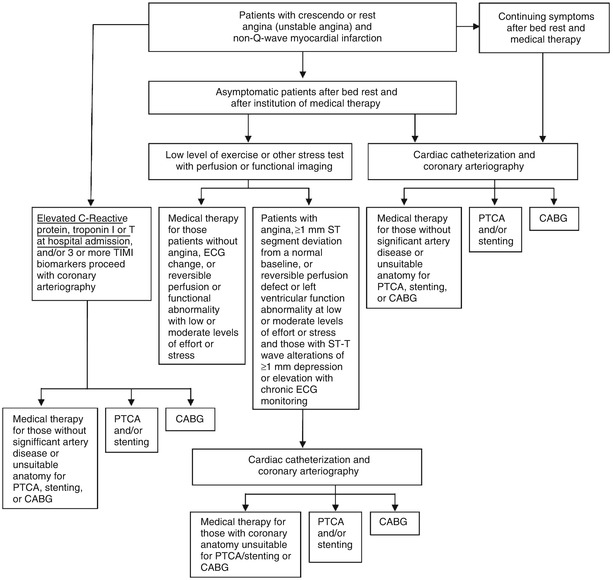
Fig. 18.1
Schematic diagram demonstrates therapeutic alternatives for the treatment of patients with unstable angina pectoris and non–ST-elevation myocardial infarction (NSTEMI). ADP adenosine diphosphate, CABG coronary artery bypass grafting, ECG electrocardiogram, PTCA percutaneous transluminal coronary angioplasty
The medical treatment regimen is intended to prevent persistent thrombus formation at the site of the unstable plaque(s) and to relieve the associated vasoconstriction. In patients without a contraindication, aspirin therapy is begun immediately. Intravenous nitroglycerin therapy is initiated, beginning at 1–2 μg/min, with dosage increases in increments of 5 μg/min every 5–10 min up to 20 μg/min to reduce systolic blood pressure to 100–120 mmHg while avoiding systemic arterial hypotension or an increased heart rate above 100 beats/min. Pain relief often occurs after complete bed rest and the institution of intravenous nitroglycerin and aspirin. Elevated blood pressure should be controlled with nitrates and an angiotensin-converting enzyme (ACE) inhibitor, and/or a beta-adrenergic-receptor blocker (beta blocker). Patients with unstable angina should be given a thrombin antagonist, unless it is contraindicated. This may be intravenous unfractionated heparin (UFH) [1], typically beginning with a bolus dose of approximately 800–1,000 units and followed by a sustained infusion of 900–1,000 U/h. The partial thromboplastin time (PTT) (or activated coagulation time [ACT]) is evaluated at 8–12-h intervals, and the heparin infusion rate is adjusted, usually on a weight-based regimen, to maintain the PTT at 55–70 s or the ACT at 250–350 s. Alternatively, low-molecular-weight heparin (LMWH) may be given subcutaneously in an appropriate dosage [10–12], i.e., 1 mg/kg body weight every 12 h. Clopidogrel (Plavix), an adenosine diphosphate (ADP) antagonist, or another ADP antagonist is additive to aspirin in preventing thrombosis and is protective in patients with acute coronary syndrome (ACS) [12, 14, 25, 28]. Clopidogrel is given as an initial loading dose of 300 mg orally followed by 75 mg per day. Alternatives to clopidogrel are prasugrel (Effient) [31–33] and ticagrelor (BRILINTA) [31, 34]. Angiotensin-converting enzyme (ACE) inhibitors or angiotensin II receptor blockers (ARBs) should be considered in the acute and chronic medical management of these patients if blood pressure and renal function allow [35]. Angiotensin II promotes inflammation and also has vasoconstrictor effects, and ACE inhibitors are likely to decrease inflammation as they protect coronary arteries [36]. However, elevated serum blood urea nitrogen (BUN) and creatinine levels may increase further with ACE therapy, especially in patients with unilateral or bilateral renal artery stenoses, limiting the utility of such therapy. Angiotensin-converting enzyme inhibitors may promote potassium retention and should be avoided in patients with hyperkalemia. Rapid lowering of total serum cholesterol and low-density lipoprotein cholesterol (LDL) levels with a statin, such as atorvastatin (Lipitor), 80 mg per day, has been shown to reduce the risk of future events in patients with ACS [37–39]. Beta blockers should be added to this therapy in patients without contraindications who have elevated blood pressure, an increased heart rate due to pain or anxiety, or complex ventricular ectopy.
Evidence-Based Overview of Specific Therapies
Several recent sets of guidelines have been published that are invaluable in the treatment of unstable angina and NSTEMI: the European Society of Cardiology (ESC) Guidelines for the Management of Acute Coronary Syndromes in Patients Presenting Without Persistent ST-segment Elevation [31] and the 2014 AHA/ACC Guideline for the Management of Patients With Non-ST-Elevation Acute Coronary Syndromes: A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines [40, 41]. Both the ESC and the AHA/ACC guidelines offer recommendations for antiplatelet and anticoagulant therapy and delineate the most recent trials upon which the evidence is based. Both include recommendations for use of the newer antiplatelet agents, prasugrel and ticagrelor. In addition, the AHA/ACC guidelines clarify, among other issues, the recommended timing, duration, and use of dual and triple antiplatelet therapy for patients with unstable angina or NSTEMI. The guidelines give evidence for the appropriate role of triple antiplatelet therapy in high-risk patients and for dual antiplatelet therapy in patients who are not at high risk. The AHA/ACC guidelines also recommend an early invasive strategy (angiography and intervention) in Class IIa patients within 24 h only for high-risk patients whose condition has been stabilized. For patients at lower risk, the guidelines allow for a more conservative strategy (Fig. 18.2 and Table 18.3).
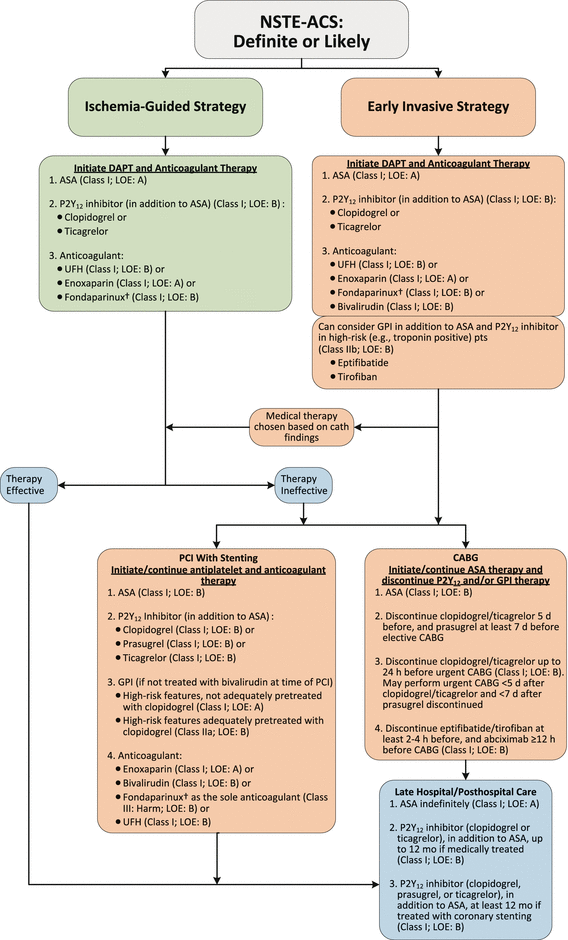
Fig. 18.2
Algorithm for management of patients with definite or likely NSTE-ACS*. *See corresponding full-sentence recommendations and their explanatory footnotes in the text of reference 39. †In patients who have been treated with fondaparinux (as upfront therapy) who are undergoing PCI, an additional anticoagulant with anti-IIa activity should be administered at the time of PCI because of the risk of catheter thrombosis. ASA indicates aspirin, CABG coronary artery bypass graft, cath catheter, COR class of recommendation, DAPT dual antiplatelet therapy, GPI glycoprotein IIb/IIIa inhibitor, LOE level of evidence, NSTE-ACS non–ST-elevation acute coronary syndrome, PCI percutaneous coronary intervention, pts patients,UFH unfractionated heparin (From Amsterdam et al. [39]. Reprinted with permission from Elsevier)
Table 18.3
Factors associated with appropriate selection of early invasive strategy or ischemia-guided strategy in patients with NSTE-ACS
Immediate invasive (within 2 h) | Refractory angina |
Signs or symptoms of HG or new or worsening mitral regurgitation | |
Hemodynamic instability | |
Recurrent angina or ischemia at rest or with low-level activities despite intensive medical therapy | |
Sustained VT or VF | |
Ischemia-guided strategy | Low-risk score (e.g., TIMI [0 or 1], GRACE [<109]) |
Low-risk Tn-negative female patients | |
Patient or clinician preference in the absence of high-risk features | |
Early invasive (within 24 h) | None of the above, but GRACE risk score >140 |
Temporal change in Tn (Section 3.4) | |
New or presumably new ST depression | |
Delayed invasive (within 25–72 h) | None of the above but diabetes mellitus |
Renal insufficiency (GFR <60 mL/min/1.73 m2) | |
Reduced LV systolic function (EF <0.40) | |
Early postinfarction angina | |
PCI within 6 months | |
Prior CABG | |
GRACE risk score 109–140; TIMI score ≥2 |
Also invaluable in the treatment of patients with unstable angina and NSTEMI are quantitative risk assessments, which are useful in clinical decision-making and triage of patients at admission for medical or interventional treatment. Of the scoring systems, the two that are the best validated and most widely used are TIMI (Thrombolysis in Myocardial Infarction) [42, 43] and GRACE (Global Registry of Acute Coronary Events) [44–46]. In direct comparison studies [47, 48] and in a recent meta-analysis of 40 derivation studies of unstable angina or NSTEMI [49], the GRACE score was most accurate both for short (in-hospital) and longer-term risk assessment, except that TIMI was more accurate for long-term outcome prediction when pro brain natriuretic peptide (BNP) was added [49]. Although derivation studies for some other scoring systems look promising, only TIMI and GRACE have been validated for all types of ACS. Compared to the TIMI score, the GRACE score is more complicated to calculate and requires a computer or other personal-digital-assistant software for determining the risk score; however, the scoring can also be done online at www.outcomes.org/grace. In addition, scoring systems have been derived to estimate levels of risk for major bleeding during hospitalization; the most widely used is the CRUSADE score (Can Rapid risk stratification of Unstable angina patients Suppress Adverse outcomes with Early implementation of the American College of Cardiology [ACC]/AHA guidelines www.crusadebleedingscore.org) [50].
Aspirin and Heparin
Taking one to four aspirin (either 81 or 325 mg) per day reduces the risk of death and MI in patients with unstable angina (Fig. 18.3) [1–3]. However, acetylsalicylic acid (ASA) may not reduce the risk of MI in healthy postmenopausal women [51, 52]. Aspirin diminishes platelet aggregation, thromboxane A2 synthesis, and inflammation and platelet–white-blood-cell interactions. The combination of aspirin and heparin inhibits the effects of thromboxane A2 and thrombin as potentiators of platelet aggregation leading to thrombosis and dynamic vasoconstriction. Inhibition of thromboxane A2 and thrombin improves regional myocardial blood flow and helps prevent thrombosis. Therefore, heparin (either UFH or LMWH) or a direct thrombin antagonist should be given to patients with unstable angina who have no contraindication. Théroux and associates [1] showed that heparin often relieves angina and reduces the risk of subsequent MI and death (Fig. 18.3); in their study, aspirin was also effective in reducing fatal and nonfatal MIs. Other researchers have shown that aspirin therapy has similar protective effects in these patients [2–4]. However, the combination of aspirin and heparin increases the risk of bleeding [1]. In patients with unstable angina or MI, abrupt withdrawal of heparin may be associated with a heparin “rebound,” entailing abrupt worsening of angina, development of MI, or both [5–7]. When heparin and other thrombin inhibitors are discontinued in these patients, it should be done slowly, over a period of several hours, with concomitant administration of aspirin and usually with other antiplatelet therapy, including clopidogrel or another adenosine diphosphate (ADP) antagonist.
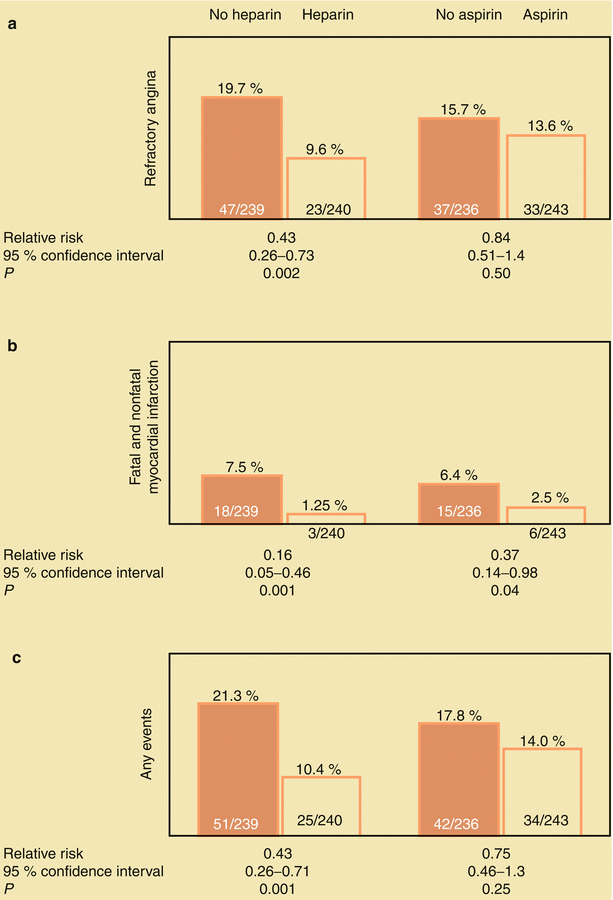
Fig. 18.3
(a) The influence of aspirin and heparin in the treatment of patients with unstable angina pectoris. Both aspirin and heparin (b) reduce the risk for fatal and nonfatal myocardial infarction (MI). Heparin also reduces the frequency of important coronary events, including death, fatal and nonfatal MI, and continuing angina. The combination of heparin and aspirin was no more successful than heparin alone in this study (Modified from Table 3 “End-point events according to treatment group” in Theroux et al. [1], with permission from Massachusetts Medical Society)
More specific thrombin inhibitors are being developed, and other pharmacologic inhibitors of thrombus are available. Low-molecular-weight heparin is a very good, and possibly preferred, alternative to UFH in patients with unstable angina and NSTEMI [8–13, 23, 24]. In these patients, disruption of an atherosclerotic plaque causes platelet activation, adhesion, and aggregation at the site of the injured plaque and activates the coagulation cascade through tissue factor release and the accumulation of multiple platelet-derived and other cell-derived mediators of thrombosis. Tissue factor complexes with factor VIIa coactivate factor Xa, thereby catalyzing the formation of thrombin (Fig. 18.4). Thrombin promotes further platelet aggregation, causes vasoconstriction at the site(s) of vascular injury, and mediates the conversion of fibrinogen to fibrin. Because both platelet activation and thrombin generation are involved in the thrombotic process, there is an obvious rationale for the use of inhibitors of both platelet aggregation and coagulation in the treatment of patients with unstable angina or NSTEMI (and patients with STEMI). Low-molecular-weight heparins act primarily through antithrombin III-mediated inhibition of factor Xa but also result in some direct thrombin inhibition. There is evidence that anti-Xa activity relates to survival and efficacy in ACS patients treated with enoxaparin [19]. Patients with low anti-Xa activity undergoing enoxaparin treatment have a lower mortality rate at 30 days if their anti-Xa activity is <0.5 IU/mL [19]. Like conventional heparin, LMWH promotes the release of tissue factor pathway inhibitor, which may contribute to the antithrombotic effects [20–22].
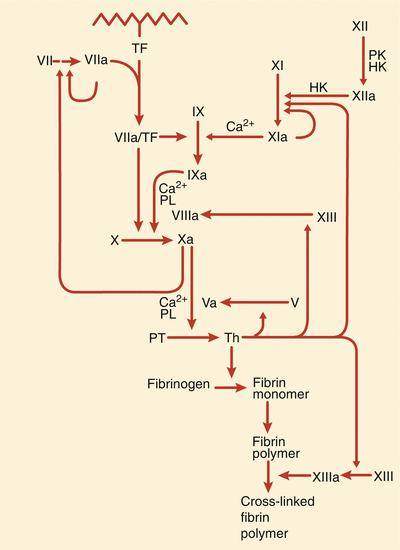
Fig. 18.4
The scheme involved in tissue factor’s (TF) participation in the coagulation cascade in the generation of thrombin (Th) and the role that thrombin plays in the development of fibrin crosslinking and platelet aggregation. HK high molecular weight kininogen, PK prekallikrein, PL phospholipid, PT prothrombin (From Loscalzo and Schafer [149]. Reproduced with permission from Wiley/Blackwell Publishing Ltd.)
Low-molecular-weight heparin is administered subcutaneously. It has a low degree of protein binding and a predictable anticoagulant response for a given dose without the need for laboratory monitoring of the PTT or ACT [20, 21]. Therefore, one can provide a consistent anticoagulant and antithrombin effect by subcutaneously injecting fixed doses of LMWH (1 mg/kg body weight), generally on a twice daily basis [20–22]. This agent is usually avoided in patients with elevated serum creatinine values (≥2.5 mg/dL) because renal insufficiency potentiates the effect of any particular dose. Instead, UFH is given to these patients. In the presence of heparin-induced thrombocytopenia (HIT) or heparin-induced thrombosis syndromes (HITS), LMWH may still be unsafe, as there is some cross-reactivity between the heparin antibody and LMWH. In this setting, if an inhibitor of thrombin is to be used, a direct-acting thrombin inhibitor is needed.
There may be advantages in continuing LMWH after hospital discharge in some patients, particularly those who do not undergo an interventional procedure, such as angioplasty or stenting, atherectomy, or surgical revascularization. There is an ongoing risk of MI or reinfarction in the subsequent 4–6 weeks in patients with unstable angina pectoris and NSTEMI; this risk is probably related to the persistent presence of endothelial injury after plaque ulceration and fissuring and the time required for its repair. The authors believe that as long as the endothelium remains anatomically disrupted or dysfunctional, the patient has a continuing risk of unstable angina and MI and that these clinical entities represent aborted STEMI with transient, rather than permanent, thrombosis. One limitation of LMWH, however, is its relatively long duration of effect (up to 24 h) after discontinuation and the absence of an effective inhibitor of that effect. In patients with bleeding problems and those requiring emergent surgery, these are important limitations.
In the late 1990s, several landmark trials—including the FRISC study (Fragmin During Instability in Coronary Artery Disease) (Fig. 18.5 and Table 18.4) [20], the TIMI II study (Thrombosis in Myocardial Infarction II) (Figs. 18.6 and 18.7) [23, 40], and the ESSENCE trial (Efficacy and Safety of Subcutaneous Enoxaparin in Non–Q-Wave coronary Events) [24] (Fig. 18.6)—showed the utility of LMWH in the treatment of patients with unstable angina and NSTEMI. These trials suggested that LMWH was clinically superior to UFH in reducing the risk of MI and death and the need for revascularization in such patients. This benefit was achieved at some increase in the risk of minor bleeding and, when continued in the outpatient phase, in minor-major bleeding. The investigators concluded that the use of LMWH should be seriously considered for these patients during the inpatient phase of therapy if they are not allergic to heparin. Figure 18.6 shows the data from five trials using LMWHs, including 21,946 patients with unstable angina and NSTEMI [8]. The ESSENCE and TIMI IIB trials were conducted in an era when clopidogrel and platelet GP IIb/IIIa antagonists were not available. Glycoprotein IIb/IIIa antagonists were protocol driven in the Acute II, Interact, and A to Z trials, and the use of clopidogrel was encouraged. All of these studies showed the noninferiority of LMWH, compared to UFH, in the treatment of patients with unstable angina and NSTEMI.
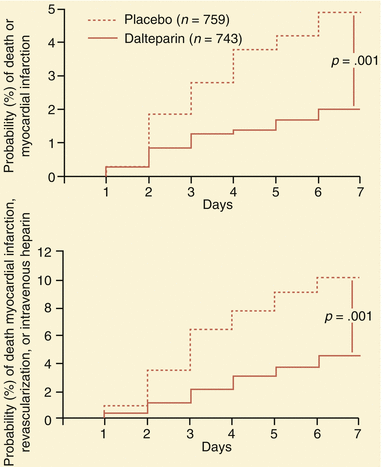
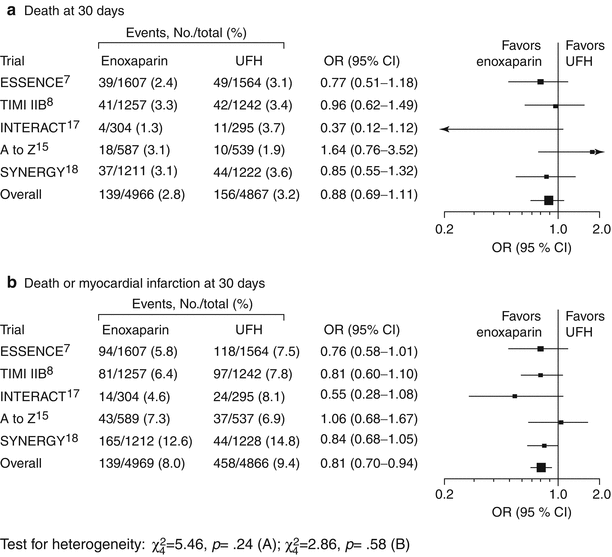
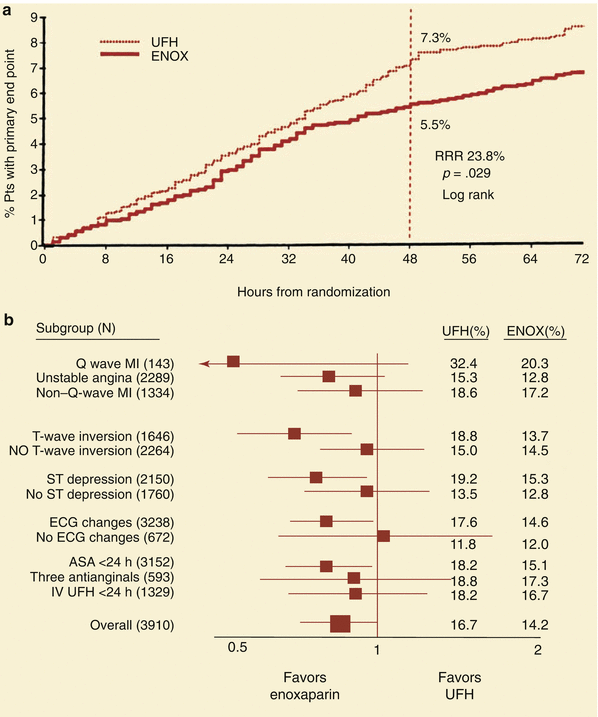
Fig. 18.5
The protective effect of low molecular weight heparin (in the form of dalteparin) in the Fragmin during Instability in Coronary Artery Disease (FRISC) study. The administration of dalteparin to patients with unstable angina and non–Q-wave myocardial infarction (MI) reduced their risk of myocardial infarction (top) and of death, MI, revascularization, or need for intravenous heparin (bottom) (see Table 18.4) (From Fragmin during Instability in Coronary Artery Disease (FRISC) study group [20]. (Reprinted with permission from Lancet Publishing Group)
Table 18.4
Absolute frequency of primary and separate endpoints for patients in the FRISC studya
Placebo (n = 749) | Dalteparin (n = 726) | Risk ratio (95 % CI) | P | |
|---|---|---|---|---|
Primary endpoints | ||||
Death or MI | 116 (15.5 %) | 102 (14.0 %) | 0.90 (0.71–1.15) | 0.41 |
Death, MI, or revascularization | 326 (43.6 %) | 296 (40.6 %) | 0.92 (0.82–1.04) | 0.18 |
Death, MI, revascularization or intravenous heparin | 337 (45.1 %) | 312 (42.7 %) | 0.94 (0.84–1.05) | 0.28 |
Exclusion of revascularization for ischemia | ||||
Death, MI, revascularization because of angina | 214 (28.7 %) | 175 (24.1 %) | 0.84 (0.70–0.99) | 0.039 |
Death, MI, revascularization because of angina or intravenous heparin | 241 (32.3 %) | 204 (28.1 %) | 0.87 (0.74–1.01) | 0.066 |
Separate endpoints | ||||
Deathb | 41 (5.5 %) | 39 (5.4 %) | 0.98 (0.64–1.50) | |
MIb | 98 (13.4 %) | 83 (11.7 %) | 0.86 (0.66–1.14) | 0.30 |
Revascularization | 254 (35.5 %) | 229 (32.9 %) | 0.92 (0.79–1.06) | 0.23 |
Revascularization because of angina | 131 (18.4 %) | 87 (12.5 %) | 0.68 (0.53–0.86) | 0.002 |
Heparin infusion | 121 (16.7 %) | 83 (11.9 %) | 0.71 (0.55–0.91) | 0.008 |
Fig. 18.6
Summary of five trials of low molecular weight heparin. Black squares indicate odds ratios (ORs); horizontal lines, 95 % confidence intervals (CIs). The size of each square reflects the statistical weight of a trial in calculating the OR, and the relative sizes of the squares are accurate within each plot only. ESSENCE Efficacy and Safety of Subcutaneous Enoxaparin in Non–Q-wave Coronary Events, TIMI IIB Thrombolysis In Myocardial Infarction IIB, ACUTE II Antithrombotic Combination Using Tirofiban and Enoxaparin II, INTERACT Integrilin and Enoxaparin Randomized Assessment of Acute Coronary Syndrome Treatment, A to Z Aggrastat to Zocor study, SYNERGY Superior Yield of the New Strategy of Enoxaparin, Revascularization and Glycoprotein IIb/IIIa Inhibitors, UFH unfractionated heparin (From Petersen et al. [8]. Reprinted with permission from American Medical Association)
Fig. 18.7
(a) The reduction in primary end point (death, MI, or urgent revascularization) in patients treated with low molecular weight heparin (ENOX, enoxaparin) compared to unfractionated heparin (UFH) in the TIMI IIB study. RRR relative risk reduction. (b) Subgroup analysis of patients in the TIMI IIB study shows that low molecular weight heparin reduces the primary end point better than unfractionated heparin (UFH) in most patient subgroups with unstable angina and non—Q-wave infarcts. ASA acetylsalicylic acid, ECG electrocardiogram, MI myocardial infarction (From Antman et al. [23]. Reprinted with permission from Wolters Kluwer Health)
Direct Thrombin Inhibitors
The direct thrombin inhibitors are useful alternatives to heparins in patients who develop thrombocytopenia or vascular thrombosis, or bleeding as an allergic response to heparin administration, i.e., HIT and HITS. The direct inhibitors of thrombin have more reliable and consistent effects on the PTT than does heparin, and patients treated with these agents do not need follow-up measurements of their PTT or ACT. The United States Food and Drug Administration (FDA) has approved four parenteral direct thrombin inhibitors—bivalirudin, lepirudin, argatroban, and desirudin—bivalirudin being the most widely used agent for patients with unstable angina/NSTEMI.
Bivalirudin
Bivalirudin (Angiomax) is a direct thrombin inhibitor that has been increasingly used in patients undergoing PCI ever since 2003, when the first bivalirudin study (the Randomized Evaluation in PCI Linking Angiomax to Reduced Clinical Events 2 [REPLACE-2 trial]), which compared heparin and a GP IIb/IIIa inhibitor with bivalirudin, suggested the noninferiority of bivalirudin therapy [53]. Later, in the ACUITY trial (Acute Catheterization and Urgent Intervention Triage Strategy), bivalirudin was superior to UFH and GP IIb/IIIa inhibition in patients with NSTEMI [54]. In this trial, approximately 40 % of the patients did not have elevated biomarkers, and more than 40 % did not have PCI. More recently, in the ISAR-REACT-4 study (Randomized Comparison of Abciximab Plus Heparin With Bivalirudin in Acute Coronary Syndrome) [55], 1,721 patients with NSTEMI were randomly assigned in a double-blind manner to receive abciximab and UFH (n = 861) or bivalirudin (n = 860). The primary end point was death, large recurrent MI, urgent target-vessel revascularization, or major bleeding within 30 days. The primary end point occurred in 10.9 % of the patients in the abciximab group and in 11 % of those in the bivalirudin group (P = 0.94) (Figs. 18.8 and 18.9) [55]. Major bleeding occurred in 4.6 % of the abciximab recipients but in only 2.6 % in the bivalirudin group (P = 0.02) (Fig. 18.8) [55]. Thus, bivalirudin treatment yielded a benefit equal to that provided by abciximab and UFH but was associated with a lower risk of major hemorrhage. In this study, bivalirudin was given as an intravenous bolus of 0.75 mg per kg body weight followed by an infusion of 1.75 mg per kg per hour for the duration of the procedure. In a meta-analysis of 3 randomized trials and 13 observational studies comparing bivalirudin versus heparin monotherapy in patients undergoing transfemoral PCI, Bertrand and coauthors [56] found equal overall outcomes with both agents but a significant decrease in major bleeding with bivalirudin (Figs. 18.10 and 18.11) [56]. However, a new substudy of ISAR-REACT-4 has suggested that platelet-function testing may be important in NSTEMI patients undergoing PCI, as patients with high platelet reactivity who underwent clopidogrel treatment had poorer outcomes than those with low platelet reactivity [57].
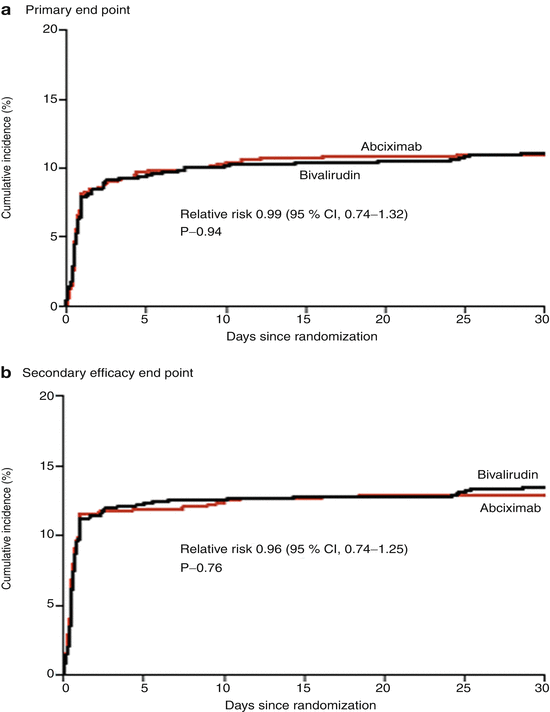
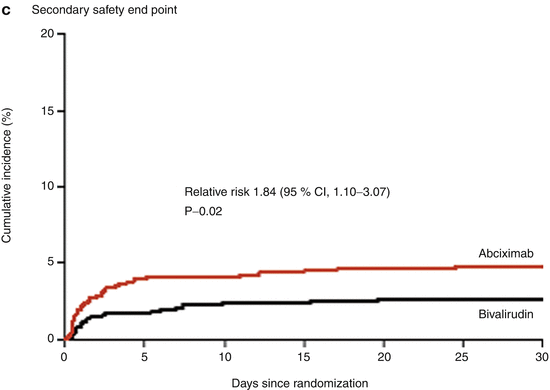
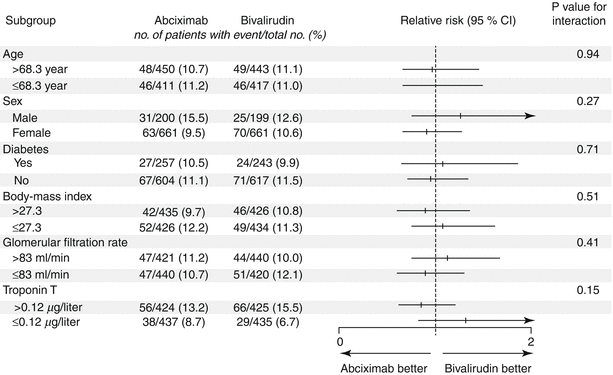
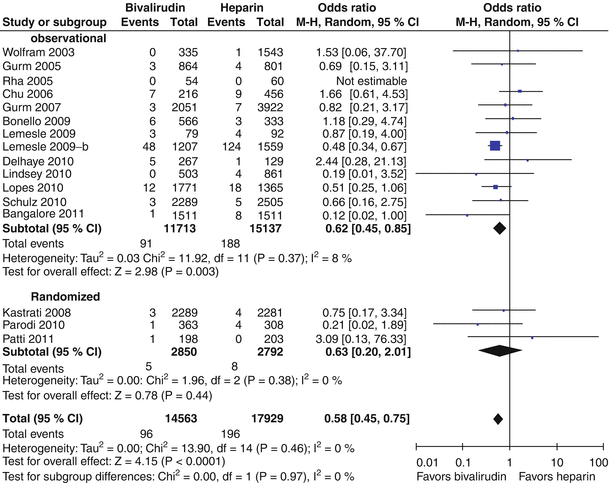
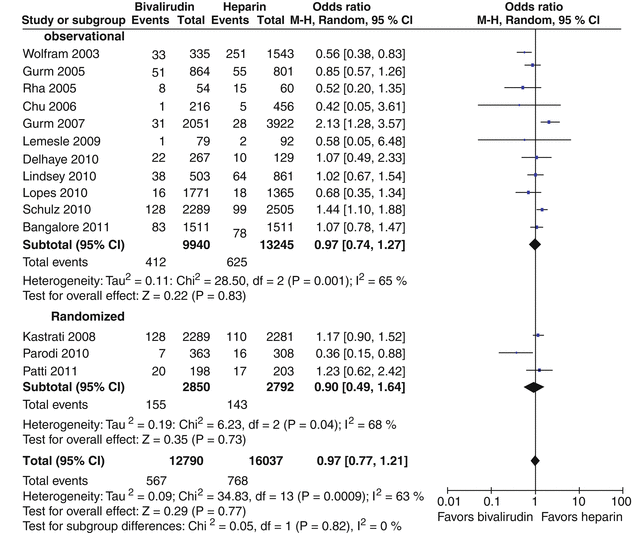
Fig. 18.8
Cumulative incidence of primary and secondary end points in two study groups comparing combination abciximab and heparin versus bivalirudin for patients with non-ST-segment elevation myocardial infarction undergoing percutaneous coronary intervention. The primary end point (a) was a composite of death, large recurrent myocardial infarction, urgent target-vessel revascularization, or major bleeding within 30 days after percutaneous coronary intervention (PCI); the secondary efficacy end point (b) was a composite of death, any recurrent myocardial infarction, or urgent target-vessel revascularization within 30 days after PCI; and the secondary safety end point (c) was major bleeding within 30 days after PCI (From Kastrati et al. [55]. Reprinted with permission from Massachusetts Medical Society)
Fig. 18.9
Thirty-day incidence and relative risk of the primary end point in prespecified subgroups. The primary end point was a composite of death, large recurrent myocardial infarction, urgent target-vessel revascularization, or major bleeding within 30 days after percutaneous coronary intervention. The body-mass index is the weight in kilograms divided by the square of the height in meters (From Kastrati et al. [55]. Reprinted with permission from Massachusetts Medical Society)
Fig. 18.10
Forest plot for incidence of death in observational studies and randomized trials. Odds ratios of individual studies (squares) and meta-analysis (diamonds) and 95 % confidence intervals (horizontal lines) are presented. M-H Mantel-Haenszel test (From Bertrand et al. [56]. Reproduced with permission of the American Journal of Cardiology. Reprinted with permission from Elsevier)
Fig. 18.11
Forest plot for incidence of myocardial infarction in observational studies and randomized trials. Odds ratios of individual studies (squares) and meta-analysis (diamonds) and 95 % confidence intervals (horizontal lines) are presented. M-H Mantel-Haenszel test (From Bertrand et al. [56]. Reprinted with permission from Elsevier)
Dabigatran
Dabigatran etexilate (Pradaxa) is an oral direct-acting thrombin antagonist that has a half-life of 12–17 h and is excreted predominantly by the kidneys. Patients receiving protein pump inhibitors may have decreased absorption. When taken with selected other medications, including amiodarone, dabigatran may result in increased absorption and increased plasma levels.
When dabigatran was used in combination with aspirin and clopidogrel in ACS patients in the RE-LY trial (Randomized Evaluation of Long-Term Anticoagulation Therapy) [58], a post hoc analysis of 6,952 patients (38.4 % of the study population) showed an overall increase in the rate of major bleeding (hazard ratio [HR] = 1.6; 95 % confidence interval [CI] = 1.41–1.81). The relative risk of increased major bleeding was similar with both dabigatran and warfarin; however, the absolute risk was lowest in the patients treated with a lower dose of dabigatran, i.e., 110 mg twice daily.
Adenosine Diphosphate Antagonists
At the site(s) of a fissured or ulcerated plaque, thrombosis may be promoted by other mediators—including ADP, serotonin, platelet-activating factor (PAF), oxygen-derived free radicals, tissue factor, and endothelin—that are not deterred by a direct thrombin inhibitor. If pain is not relieved with bed rest and the use of intravenous nitrates, aspirin, and a heparin, the risk of subsequent MI, sudden death, and ventricular arrhythmias is increased. Platelet-aggregation inhibitors that are more comprehensive than aspirin and heparin, decreasing platelet aggregation in response to most or all of the above-named mediators, can be useful in the treatment of such patients; these agents include an ADP antagonist, such as clopidogrel [14], prasugrel [31, 32], or ticagrelor [31, 59], and/or an inhibitor of platelet GP IIb/IIIa receptors, including the monoclonal antibody abciximab (ReoPro), the synthetic peptide inhibitor eptifibatide (Integrilin), or a low-molecular-weight inhibitor, such as tirofiban (Aggrastat) [15–18]. A platelet GP IIb/IIIa receptor antagonist (discussed in detail later in this chapter) may be added to this therapeutic regimen for patients who continue to have resting angina and who will undergo interventional therapy. The platelet GP IIb/IIIa inhibitors seem especially useful in patients with diabetes mellitus [18] and in those with elevated serum troponin levels who will undergo PCI [15–17]. If resting angina persists despite the medical therapy outlined above, one should consider adding intraaortic balloon counterpulsation (Fig. 18.12), which almost always relieves rest pain in patients with unstable angina or NSTEMI. Patients who have continuing angina at rest and require intensive combined therapy must urgently proceed to coronary arteriography and PCI or coronary artery bypass grafting surgery (CABG). (See also sections “Interventional therapy” and “General considerations.”)
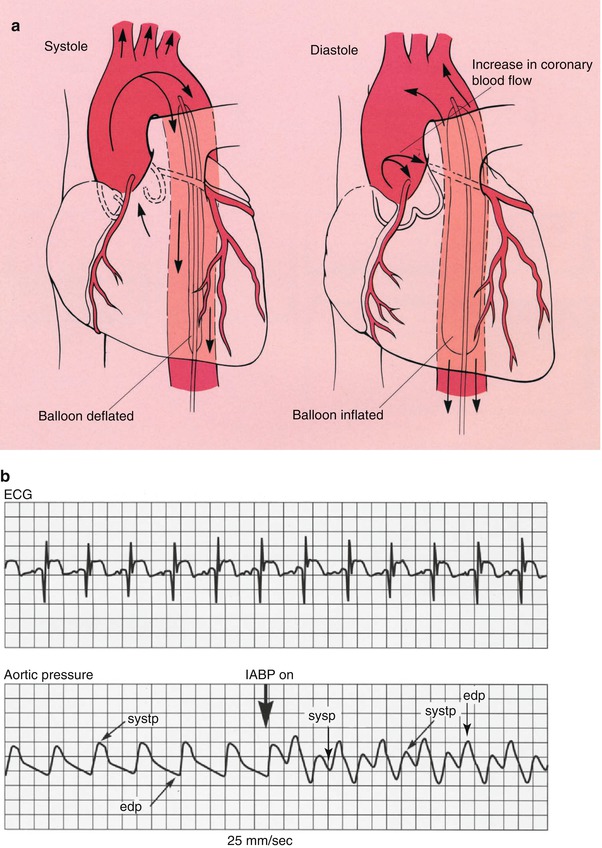
Fig. 18.12
(a) The position in which the intraaortic balloon pump (IABP) is placed in the aorta. The balloon deflates during cardiac systole and inflates during cardiac diastole. The net results of this filling and collapsing action are to increase the diastolic blood pressure in the proximal aorta and consequently to increase coronary blood flow. The systolic collapse of the balloon reduces the work of the heart. (b) The physiologic effects of the IABP. The increase in augmented diastolic blood pressure (diastp) associated with balloon inflation (IABP on) is shown. The electrocardiogram (ECG) is used to time cardiac systole and diastole. edp end diastolic pressure, sysp systolic blood pressure (From Willerson [156]. Reprinted with permission from Gower Medical Publishing)
Clopidogrel
As noted earlier, clopidogrel (Plavix) blocks ADP receptors, thus inhibiting platelet aggregation. Less well recognized is the fact that clopidogrel also reduces platelet aggregation to thromboxane A2 and serotonin [26]. Thus, clopidogrel exerts a powerful inhibitory effect on platelet aggregation at sites of atherosclerotic plaque fissuring and ulceration.
Table 18.5 summarizes the CURE trial (Clopidogrel in Unstable angina to Prevent Recurrent Events) [28] and the CREDO trial (Clopidogrel for the Reduction of Events During Observation) [60], both of which evaluated the effects of clopidogrel in patients with ACS. CURE was the first to show that combined aspirin and clopidogrel therapy reduced the incidence of long-term thrombotic events. In this trial, 2,658 patients (21 %) underwent PCI a median of 10 days after enrollment. The timing was decided by physician preference rather than trial design. The patients had a relative risk reduction of 30 % for cardiovascular death, MI, or urgent revascularization from the time of the PCI procedure through day 30 (P = 0.03) [27, 28, 61]. There was an approximately 20 % reduction (9.3 % of patients in the clopidogrel group versus 11.4 % of patients in the placebo group) in the risk of vascular death, MI, and stroke after 9 months of therapy with both aspirin and clopidogrel (Fig. 18.13; Table 18.5) [27, 28]. From the PCI procedure through the remainder of the 12-month follow-up period, there was a relative risk reduction of 25 % (P = .047) [27, 28, 61]. Subsequently, CREDO [60] showed that long-term therapy with clopidogrel and aspirin significantly reduced the risk of adverse ischemic events after elective PCI.
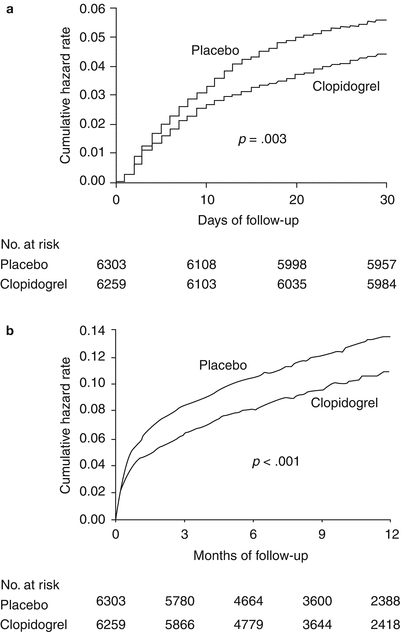
Table 18.5
Clinical trials with clopidogrel in patients with acute coronary syndrome (ACS) and those with elective percutaneous coronary intervention (PCI)
Trial | No. of patients | Primary outcome | Duration of follow-up (months) | Results |
|---|---|---|---|---|
1. CURE (Circulation. 2003; 107:966–972 and N Engl J Med. 2001;345:494–502) | 12,562 | Cardiovascular death, myocardial infarction, or stroke | 12 | 30 % relative risk reduction; 25 % relative risk reduction for those receiving PCI |
2. CREDOa (JAMA. 2002; 288:2411–2420) | 2,116 patients with PCI, with or without a stent | Cardiovascular death, myocardial infarction, or stroke | 12 | 27 % reduction in the composite end point |
Fig. 18.13
Cumulative hazard rates for death from cardiovascular causes, nonfatal MI, and stroke, in patients presenting with acute coronary syndromes without ST-T segment elevation in the CURE trial. (a) Days of follow-up. (b) Months of follow-up (From Yusuf et al. [28]. Reprinted with permission from Massachusetts Medical Society)
Yusuf and colleagues [27] studied the rapidity with which treatment in the CURE trial was effective and the sustainability of that treatment over 1 year, as well as the rates of bleeding during the same period. A total of 12,562 patients with ACS were randomized to receive clopidogrel (300 mg initially, followed by 75 mg per day orally) or placebo for 3–12 months. All patients were also receiving aspirin therapy. The incidence of cardiovascular death, MI, or stroke at 30 days was 5.4 % in the placebo group and 4.3 % in the actively treated group (relative risk, 0.79). After 36 days, the respective rates were 6.3 % versus 5.2 % (relative risk, 0.82). No significant increase in life-threatening bleeding was observed at either 30 days or 12 months, but clopidogrel-treated patients had increased minor and major bleeding and needed more blood transfusions. According to the early treatment data, a benefit was documented within 24 h of treatment, with consistently lower rates of adverse outcomes.
More recently, in the CURRENT-OASIS 7 trial [62], 25,086 ACS patients undergoing invasive treatment were randomized to receive either a high dose or a standard dose of clopidogrel. The high-dose group received a 600-mg loading dose of clopidogrel and then 150 mg once daily for next 7 days, followed by 75 mg once daily until 30 days. The standard-dose group received a 300-mg loading dose of clopidogrel, followed by 75 mg once daily until 30 days. Patients also were given 300–325 mg of aspirin once daily or 75–100-mg aspirin once daily. No significant intergroup difference was found in the primary efficacy end point, but the higher clopidogrel dose was associated with increased bleeding. There was no significant difference between the higher and lower doses of aspirin with respect to the primary outcome or major bleeding.
Thus, clopidogrel should be added to aspirin, nitrates, and a thrombin inhibitor (a heparin) in the immediate and long-term therapy of ACS. In patients selected for elective or semielective CABG, clopidogrel should be discontinued for at least 5 days in advance of surgery, with demonstration of a return to relatively normal platelet aggregation in response to ADP before CABG. Otherwise, there is a major risk of bleeding intraoperatively and postoperatively. Major bleeding associated with clopidogrel treatment is treated with multiple infusions of fresh platelets. Because the effects of clopidogrel may be somewhat reduced in patients with diabetes, these patients may benefit from one of the more potent antiplatelet agents [63].
One issue that is not yet completely resolved is how effective and safe clopidogrel is when added to the treatment of ACS patients who are already receiving aspirin, a heparin, and a GP IIb/IIIa receptor antagonist. Similarly unclear is how much additional protective benefit versus risk is conferred by a GP IIb/IIIa antagonist in patients who are already receiving clopidogrel, aspirin, and heparin. These issues may be answered in future clinical trials, but at this time ACS patients receiving aspirin, clopidogrel, heparin, and nitrates should probably have a GP IIb/IIIa antagonist added only if they continue to have resting angina and will have a PCI or if they have complex coronary artery anatomy or diabetes mellitus and are undergoing PCI. Both clopidogrel and GP IIb/IIIa antagonists need to be withdrawn before CABG if at all possible; otherwise, the patient will have a substantial intraoperative and postoperative bleeding risk that usually requires many units of platelets to overcome—if it can be overcome at all.
Some researchers have reported that the combined administration of atorvastatin (Lipitor) and clopidogrel reduces clopidogrel’s ability to inhibit platelet aggregation in response to ADP, as a result of clopidogrel’s being metabolized by the cytochrome P4503A4 system, which might then competitively inhibit the metabolic activation of clopidogrel in the liver [37, 64]. However, investigators in the CREDO trial were unable to show an inhibiting effect of atorvastatin in a placebo-controlled post hoc analysis [39]. If there is an important clinical interaction between these drugs in vivo, it is probably limited to a relatively small number of genetically susceptible individuals. For more information about the use of statins in patients with unstable angina or NSTEMI, see the section “Acute Lipid Lowering with a Statin.”
In reducing major adverse cardiac events, clopidogrel is at least as efficacious as ticlopidine (Ticlid) and has better tolerability and fewer side effects, including a much lower hematologic risk. Because ticlopidine has been associated with significant and sometimes fatal adverse reactions, including neutropenia and bone marrow aplasia, it should be replaced by clopidogrel [65, 66].
Prasugrel
Prasugrel (Effient) is a novel thienopyridine and a prodrug that, like clopidogrel, requires conversion to an active metabolite before binding to the platelet P2Y12 receptor. Prasugrel inhibited ADP-induced platelet aggregation more rapidly and more consistently than did standard and higher doses of clopidogrel in healthy volunteers [67] and in patients with coronary heart disease [68]. TRITON-TIMI 38 (Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition with Prasugrel Thrombolysis in Myocardial Infarction) was a pivotal trial for prasugrel, which was approved by the FDA in 2009. In TRITON-TIMI 38, 13,608 patients with ACS and scheduled PCI were randomized to receive a loading dose of 60 mg of prasugrel or 300 mg of clopidogrel [69, 70]. The series included 10,074 patients with unstable angina or NSTEMI and 3,534 patients with STEMI. After PCI, the patients received maintenance doses of either 10 mg of prasugrel or 75 mg of clopidogrel daily. Use of aspirin was required. The primary efficacy end point was death from cardiovascular causes, nonfatal MI, or nonfatal stroke. The primary end point occurred in 12.1 % of patients receiving clopidogrel and 9.9 % of patients receiving prasugrel (P < 0.001) (Fig. 18.14) [69]. The prasugrel-treated patients also had significant reductions in the rates of MI (9.7 % for clopidogrel vs. 7.4 % for prasugrel [P < 0.001]); urgent target-vessel revascularization (3.7 % vs. 2.5 %; P < 0.001); and stent thrombosis (2.4 % vs. 1.1 %; P < 0.001). However, 2.4 % of patients receiving prasugrel and 1.8 % of patients receiving clopidogrel (P = 0.03) experienced major bleeding (Table 18.6). The rates of both nonfatal bleeding (1.1 % vs. 0.9 %; P = 0.23) and fatal bleeding (0.4 % vs. 0.1 % P = 0.002) were greater in the prasugrel-treated patients. Thus, compared to the clopidogrel group, prasugrel recipients with ACS undergoing PCI had a reduction in ischemic events, but an increased risk of major bleeding, suggesting that a greater level of ADP antagonism provides increased protection from ischemic events, but a greater likelihood of bleeding. There was no associated mortality benefit for PCI. There was a lower rate of death after CABG as compared to clopidogrel, despite an increase in bleeding, platelet transfusion, and surgical re-exploration for bleeding [71]. In a comparison of prasugrel to clopidogrel in patients under age 75 with ACS who did not undergo revascularization, prasugrel was not associated with a significant decrease in the primary endpoint: death from cardiovascular causes, MI, or stroke (prasugrel, 13.9 %; clopidogrel, 16 % [hazard ratio for clopidogrel, 0.91; 95 % confidence interval]) [72]. The risk of severe and intracranial bleeding was similar between the 2 groups, but, as could be expected, higher rates of minor or moderate pleading occurred in the prasugrel group.
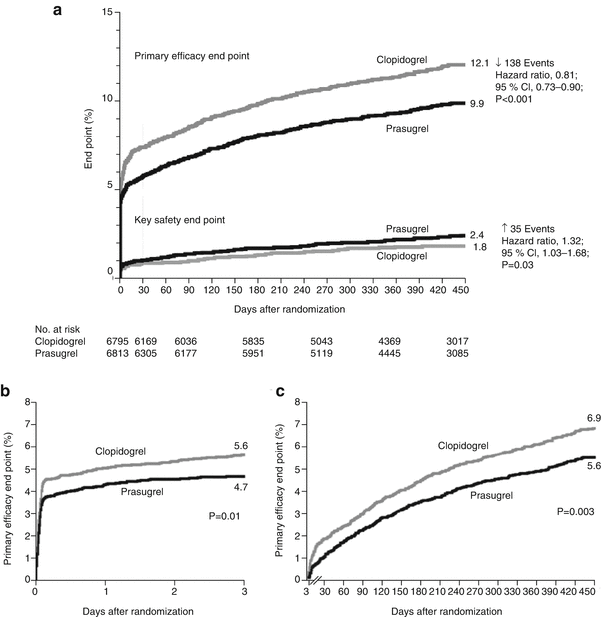
Fig. 18.14
Cumulative Kaplan-Meier estimates of the rates of key study end points during the follow-up period. (a): Data for the primary efficacy end point (death from cardiovascular causes, nonfatal myocardial infarction [MI], or nonfatal stroke) (top) and for the key safety end point (TIMI major bleeding not related to coronary artery bypass grafting) (bottom) during the full follow-up period. The hazard ratio for prasugrel, as compared with clopidogrel, for the primary efficacy end point at 30 days was 0.77 (95 % confidence interval [CI], 0.67–0.88; P < 0.001) and at 90 days was 0.80 (95 % CI, 0.71–0.90; P < 0.001). (b): Data for the primary efficacy end point are also shown from the time of randomization to day 3. (c): Data from 3 days to 15 months, with all end points occurring before day 3 censored. (c): The number at risk includes patients who were alive (regardless of whether a nonfatal event had occurred during the first 3 days after randomization). The P values in (a) for the primary efficacy end point were calculated with the use of the Gehan-Wilcoxon test. All other P values were calculated with the use of the log-rank test [69]
Table 18.6
Thrombolysis in myocardial infarction (TIMI) bleeding end points in the overall cohort at 15 months
End point | Prasugrel (N = 6,741) | Clopidogrel (N = 6,716) | Hazard ratio for prasugrel (95 % CI) | P value |
|---|---|---|---|---|
No. of patients (%) | ||||
Non–CABG-related TIMI major bleeding (key safety end point) | 146 (2.4) | 111 (1.8) | 1.32 (1.03–1.68) | 0.03 |
Related to instrumentation | 45 (0.7) | 38 (0.6) | 1.18 (0.77–1.82) | 0.45 |
Spontaneous | 92 (1.6) | 61 (1.1) | 1.51 (1.09–2.08) | 0.01 |
Related to trauma | 9 (0.2) | 12 (0.2) | 0.75 (0.32–1.78) | 0.51 |
Life-threateninga | 85 (1.4) | 56 (0.9) | 1.52 (1.08–2.13) | 0.01 |
Related to instrumentation | 28 (0.5) | 18 (0.3) | 1.55 (0.86–2.81) | 0.14 |
Spontaneous | 50 (0.9) | 28 (0.5) | 1.78 (1.12–2.83) | 0.01 |
Related to trauma | 7 (0.1) | 10 (0.2) | 0.70 (0.27–1.84) | 0.47 |
Fatalb | 21 (0.4) | 5 (0.1) | 4.19 (1.58–11.11) | 0.002 |
Nonfatal | 64 (1.1) | 51 (0.9) | 1.25 (0.87–1.81) | 0.23 |
Intracranial | 19 (0.3) | 17 (0.3) | 1.12 (0.58–2.15) | 0.74 |
Major or minor TIMI bleeding | 303 (5.0) | 231 (3.8) | 1.31 (1.11–1.56) | 0.002 |
Bleeding requiring transfusionc | 244 (4.0) | 182 (3.0) | 1.34 (1.11–1.63) | <0.001 |
CABG-related TIMI major bleedingd | 24 (13.4) | 6 (3.2) | 4.73 (1.90–11.82) | <0.001 |
In addition, except in high-risk patients (i.e., those with diabetes or a history of a previous MI), prasugrel is not recommended for persons 75 years of age or older, because it has an uncertain benefit in this age group and an increased risk of causing a possibly fatal intracranial hemorrhage [40].
Ticagrelor
Recent clinical guidelines suggest that dual antiplatelet therapy should be prescribed for patients with ACS. However, as mentioned earlier, clopidogrel is a prodrug that has to be converted to an active drug capable of inhibiting platelet aggregation. Prasugrel increases antiplatelet activity but also increases the risk of major bleeding [69]. Another and possibly better option than prasugrel is ticagrelor (BRILINTA). Ticagrelor is a reversible and direct-acting oral ADP inhibitor that has shown consistent increased P2Y12 inhibition. It has a shorter half-life than thienopyridines—including ticlopidine, clopidogrel, and prasugrel—and does not need to undergo metabolic activation via the liver (Fig. 18.15) [34].
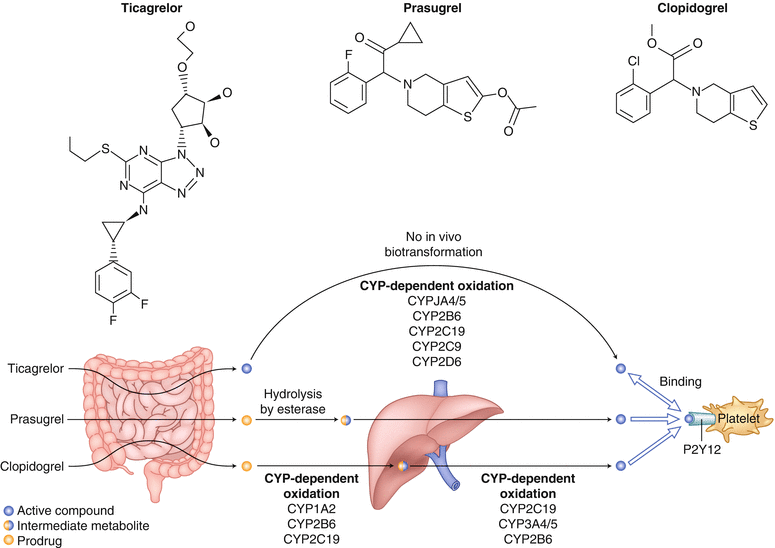
Fig. 18.15
Biotransformation and mode of action of clopidogrel, prasugrel, and ticagrelor. Ticagrelor, a cyclopentyl triazolopyrimidine, is rapidly absorbed in the intestine. The absorbed drug directly and reversibly binds to the platelet adenosine diphosphate (ADP) receptor, P2Y12, and does not require further biotransformation for activation. The half-life of ticagrelor is 7–8 h. The thienopyridines, prasugrel and clopidogrel, are pro-drugs. Their active metabolites irreversibly bind to P2Y12 for the platelet’s life span. After intestinal absorption of clopidogrel, two cytochrome P-450 (CYP)-dependent oxidation steps are required to generate active compound. After prasugrel is absorbed in the intestine, it is rapidly hydrolyzed by means of esterases to an intermediate metabolite, and one further CYP-dependent oxidation step is required to generate active compound. CYP isoenzymes involved in the activation of both clopidogrel and prasugrel are also shown. Their activity may be affected by genetic polymorphisms. (From Schomig [157]. Reprinted with permission of Massachusetts Medical Society)
The PLATO trial (PLATelet inhibition and patient Outcomes) [73–78], a multicenter, double-blind, randomized study, evaluated ticagrelor (in a 180-mg loading dose followed by 90 mg twice daily thereafter) versus clopidogrel (in a 300–600-mg loading dose followed by 75 mg daily thereafter) for the prevention of cardiovascular events in 18,624 patients with ACS, with or without ST-segment elevation (Figs. 18.16 and 18.17 [70] and Tables 18.7 and 18.8 [34]). At 12 months, the primary end point, a composite of vascular-related death, MI, or stroke, had occurred in 9.8 % of the ticagrelor recipients versus 11.7 % of the clopidogrel group (P < 0.001) (Fig. 18.16) [73]. Moreover, ticagrelor significantly decreased the incidence of MI alone (5.8 % vs. 6.9 %; P = 0.005) and death from vascular causes (4.0 % vs. 5.1 %, P = 0.001) but not stroke alone (1.5 % vs. 1.3 %, P = 0.22). The incidence of death from any cause was also reduced with ticagrelor (4.5 % vs. 5.9 % with clopidogrel). However, there was no significant difference in the rates of major bleeding between the two groups (11.6 % with ticagrelor vs. 11.2 % with clopidogrel) (Fig. 18.17) [73]. Ticagrelor was associated with a higher rate of major bleeding not related to CABG (4.5 % vs. 3.8 %; P = 0.03), including more instances of fatal intracranial hemorrhage and fewer fatal bleeding episodes of other types. Thus, in ACS patients with and without ST-segment elevation, ticagrelor reduced the rate of death from vascular causes, MI, and stroke compared to clopidogrel, without an increase in the rate of overall major bleeding but with an increase in the rate of non–procedure-related hemorrhage, including fatal intracranial bleeding [69]. In the PLATO study, the ticagrelor-treated patients more frequently had certain other adverse effects, including dyspnea (13.8 % vs. 7.8 % with clopidogrel), although few patients discontinued the study because of dyspnea (0.9 % vs. 0.1 % [clopidogrel]). Discontinuation of the study because of adverse events occurred more frequently with ticagrelor than with clopidogrel (7.4 % vs. 6 %; P < 0.001).
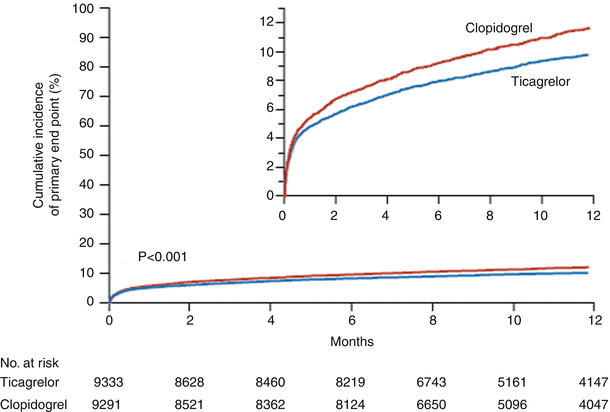
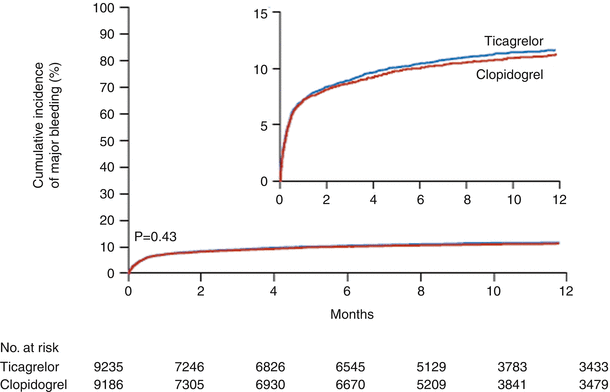
Fig. 18.16
Cumulative Kaplan-Meier estimates of the time to the first adjudicated occurrence of the primary efficacy end point. The primary end point—a composite of death from vascular causes, myocardial infarction, or stroke—occurred significantly less often in the ticagrelor group than in the clopidogrel group (9.8 % vs. 11.7 % at 12 months; hazard ratio, 0.84; 95 % confidence interval, 0.77–0.92; P < 0.001) (From Wallentin et al. [73]. Reprinted with permission from Massachusetts Medical Society)
Fig. 18.17
Cumulative Kaplan-Meier estimates of the time to the first major bleeding end point, according to the study criteria. The time was estimated from the first dose of the study drug in the safety population. The hazard ratio for major bleeding, defined according to the study criteria, for the ticagrelor group as compared with the clopidogrel group was 1.04 (95 % confidence interval, 0.95–1.13) (From Wallentin et al. [73]. Reprinted with permission from Massachusetts Medical Society)
Table 18.7
Comparison of main properties of antiplatelet agents
Ticagrelor | Prasugrel | Clopidogrel | Aspirin | |
|---|---|---|---|---|
Basic information | New class of orally active nonthienopyridine antiplatelet agents, the cyclopentyl triazolopyrimidines | Third generation thienopyridine | Second generation thienopyridine | Aspirin has an antiplatelet effect by inhibiting the production of thromboxane, which binds platelet molecules together |
Mechanism of action | Reversible inhibitor of the adenosine diphosphate (ADP) P2Y12 receptor | Irreversible inhibitor of the adenosine diphosphate (ADP) P2Y12 receptor | Irreversible inhibitor of the adenosine diphosphate (ADP) P2Y12 receptor | Suppresses the production of prostaglandins and thromboxanes, owing to its irreversible inactivation of the cyclooxygenase (COX) enzyme |
Active substance drug/metabolite | Both drug and its metabolite are active | Drug is inactive and needs to be metabolized to active metabolites | Drug is inactive and needs to be metabolized to active metabolites | Acetylsalicylic acid |
Time to achieve maximum platelet inhibition or maximum plasma concentration | After loading dose of 180 mg, the maximum plasma concentrations and maximum platelet inhibition are reached in 1–3 h | After loading dose of 60 mg, the maximum 60–70 % platelet inhibition is usually achieved in 2–4 h, maximum plasma concentration of active metabolite is reached within 0.5 h | After loading dose of 600 mg, the maximum plasma concentration is achieved in 1 h and maximum platelet inhibition is within 2–3 h | Plasma concentration is dose dependent, can continue to rise for up to 24 h after ingestion when overdose |
The mean elimination half-life | The mean elimination half-life is 6–13 h (dose independent) | The mean elimination half-life of active metabolite is 3.7 h | After a single dose of 75 mg, half-life is approximately 6 h. The elimination half-life of the inactive acid metabolite is 8 h after a single and repeated dose | Elimination half-life of about 2–4.5 h. When higher doses of salicylate are ingested (more than 4 g), the half-life becomes much longer (15–30 h) |
Absorption | Rapidly absorbed | Rapidly absorbed | Rapidly absorbed | Acetylsalicylic acid is poorly soluble in the acidic conditions of the stomach, which can delay absorption of high doses for 8–24 h |
Elimination | No specific data have been found. | Approximately 70 % of prasugrel metabolites eliminated by kidney | Approximately 40 % of a 75 mg dose is excreted in urine and 35–60 % is excreted in feces | Salicylates are excreted mainly by the kidneys as salicyluric acid (75 %), free salicylic acid (10 %), salicylic phenol (10 %) and acyl (5 %) glucuronides, and gentisic acid ( <1 %) |
Table 18.8
Three randomized trials of adenosine diphosphate receptor antagonists in acute coronary syndromes
PLATO | TRITON- TIMI 38 | CURE | |
|---|---|---|---|
Number of patients | 18,624 | 13,608 | 12,562 |
Drugs tested | Ticagrelor vs clopidogrel | Prasugrel vs clopidogrel | Placebo vs clopidogrel
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|