Left ventricular (LV) Systolic wall stress
LV systolic pressure
LV diameter
LV wall thickness
Contractility
Heart rate
Systolic Wall Tension
The force produced by the contracting myocardium to generate pressure is expressed in terms of wall tension. Systolic wall tension is directly proportional to left ventricular systolic pressure. Systolic wall tension is expressed as force per unit cross-sectional area, and so is directly proportional to left ventricular cavity diameter and inversely proportional to wall thickness. Since wall tension cannot be directly measured, systolic arterial pressure is used as a surrogate for wall tension. Because the effects of left ventricular cavity diameter and wall thickness are neglected, the relationship between systolic arterial pressure and myocardial O2 consumption is imprecise. However, changes in systolic wall stress are directly proportional to changes in systolic blood pressure and are indicative of changes in O2 demands.
Contractility
Myocardial contractility describes the rate of tension development during cardiac contraction. An increase in contractility causes an increase in the rate of left ventricular pressure development and an increase in the velocity of shortening of the contracting myocardium. The first time derivative of left ventricular pressure during isovolumic contraction (dP/dtmax) can be used to estimate myocardial contractility. This variable changes in parallel with contractility, but also can be influenced by both left ventricular preload (end-diastolic volume) and afterload (systolic pressure). This index of contractility can be determined from high fidelity recording of left ventricular pressure or estimated from the early velocity curve of the mitral regurgitation spectral profile on echo Doppler recordings.
Heart Rate
Heart rate is a summing factor for the energy cost of cardiac contraction. Since systolic wall tension and contractility are computed on a per beat basis, changes in heart rate bear a strong relationship to changes in myocardial O2 demand. Furthermore, heart rate directly influences contractility, so that increases of heart rate during exercise or other stress result in increased contractility.
Rate-Pressure Product
The product of heart rate and systolic arterial pressure (rate- pressure product) is a convenient index for estimating changes in myocardial O2 demands. Proportional changes in heart rate or systolic pressure have equal effects on myocardial O2 consumption [5]. The rate-pressure product is especially useful for estimating the cardiac workload during exercise testing.
Epicardial Coronary Arteries
The epicardial arteries arborize over the surface of the heart and give off branches that penetrate into the myocardium. Normal epicardial arteries are true conduit vessels that contribute little to total coronary vascular resistance. The epicardial arteries are richly innervated with sympathetic nerve fibers, and coronary artery smooth muscle cells contain both α-adrenoceptors which mediate vasoconstriction as well as β-receptors that cause vasodilation [6]. In the normal heart exercise results in endothelium-dependent vasodilation of the epicardial arteries which is mediated by nitric oxide, so that blockade of nitric oxide production prevents normal coronary artery vasodilation during exercise [7, 8]. Similarly, endothelial dysfunction resulting from hyperlipidemia, hypertension or atherosclerosis blunts or abolishes epicardial artery dilation during exercise [9].
Autopsy studies of patients with atherosclerotic disease demonstrate that approximately 75 % of coronary atheromas are eccentric in location, leaving part of the vessel wall relatively uninvolved. The existence of a relatively uninvolved segment explains the observation that coronary stenoses often do not behave as fixed narrowings, but have some degree of compliance and are able to undergo active vasomotion [10]. The intraarterial distending pressure within a stenosis opposes the vasomotor tone and elasticity of the arterial wall which act to constrict the vessel. Compliant stenoses interact with the distal resistance vessels as the result of hydrodynamic changes in the stenotic segment. Thus, when exercise results in vasodilation of the distal resistance vessels, the increased blood velocity (kinetic energy) within the stenotic segment causes a proportionate decrease in pressure (potential energy) acting to distend the stenosis. As the distending pressure in the stenosis decreases with increasing flow, the arterial wall elasticity and vasoconstrictor tone act to collapse the stenosis and increase stenosis severity [11]. This effect is augmented by endothelial dysfunction in the atherosclerotic coronary circulation, since the normal flow-mediated vasodilator response is absent [12]. Furthermore, a compliant segment of arterial wall at the site of a stenosis can undergo active vasoconstriction in response to sympathetic nervous system activation or agonists such as ergonovine, serotonin or thromboxane. Sympathetic vasoconstriction is augmented in the atherosclerotic coronary circulation, since alpha2-adrenergic stimulation normally causes endothelial release of nitric oxide which opposes vasoconstriction in normal coronary vessels but is lost in atherosclerotic vessels [13]. Isometric exercise causes greater activation of the sympathetic nervous system than dynamic exercise; consequently, the tendency toward constriction of stenotic coronary artery segments is more prominent with isometric exercise than with dynamic exercise [10].
Coronary Resistance Vessels
The principal resistance to coronary blood flow resides in microvessels smaller than 400 μm in diameter. Coronary resistance vessels can be divided functionally into two separate segments: arterioles and resistance arteries. Coronary arterioles less than 100 μm in diameter mediate metabolic vasoregulation and autoregulation, by which blood flow is adjusted in response to myocardial demands and is maintained constant over a range of perfusion pressures. Coronary resistance arteries 100–300 μm in diameter account for as much as 40 % of total coronary resistance but are not responsive to the metabolic needs of the myocardium [14]. Because of important differences in the responses of the coronary arterioles and the resistance arteries, these vascular segments will be discussed separately.
Coronary Arterioles
Regulation of blood flow in response to changing myocardial needs occurs at the level of the coronary arterioles (Fig. 4.1). Adenosine has been suggested as a mediator that couples myocardial metabolism to coronary resistance vessel activity. When cardiac work is increased, the rate of ATP utilization by the contractile apparatus transiently exceeds the rate of resynthesis of ATP, resulting in an increase of free ADP. The cytosolic enzyme adenylate kinase can then act on two molecules of ADP to form one molecule each of ATP and AMP. AMP released by this reaction can be catabolized to adenosine which can be transported out of the myocyte into the interstitial fluid. In the interstitial fluid adenosine engages A2A receptors on the coronary smooth muscle cell membrane to cause vasodilation [15]. Adenosine has a half-life of seconds, being rapidly reincorporated into the adenine nucleotide pool or deaminated to inosine, which has little vasodilator activity. The vasodilator effect of adenosine can be inhibited by methyl xanthines such as theophylline which act as adenosine receptor blockers. Adenosine receptor blockade does not interfere with the normal increase in myocardial blood flow that occurs during exercise or other increases of O2 demands, implying that adenosine is not a mediator of normal metabolic vasoregulation [16]. However, adenosine production increases markedly during ischemia and does contribute to vasodilation of coronary resistance vessels in ischemic myocardial regions [16]. Other metabolic perturbations that occur during ischemia, including accumulation of hydrogen ion and lactate, also cause coronary vasodilation, but these changes are not likely to contribute to regulation of arteriolar tone during physiologic conditions. Decreases of arterial O2 tension and increases of carbon dioxide tension exert vasodilator effects on coronary resistance vessels, with evidence for a synergistic interaction between them. Oxygen diffusion out of the coronary microvessels occurs so rapidly that O2 tension in arterioles and even small arteries is lower than in aortic blood, suggesting a direct role for O2 in the local regulation of blood flow [17]. There is evidence erythrocytes not only act as O2 carriers, but also contribute to metabolic vasoregulation by releasing ATP at low O2 tensions which causes endothelium dependent vasodilation of the coronary resistance vessels [18]. Thus, regulation of blood flow in response to changes of cardiac work and oxygen needs is dependent on multiple metabolic messengers that act on the coronary resistance vessels.
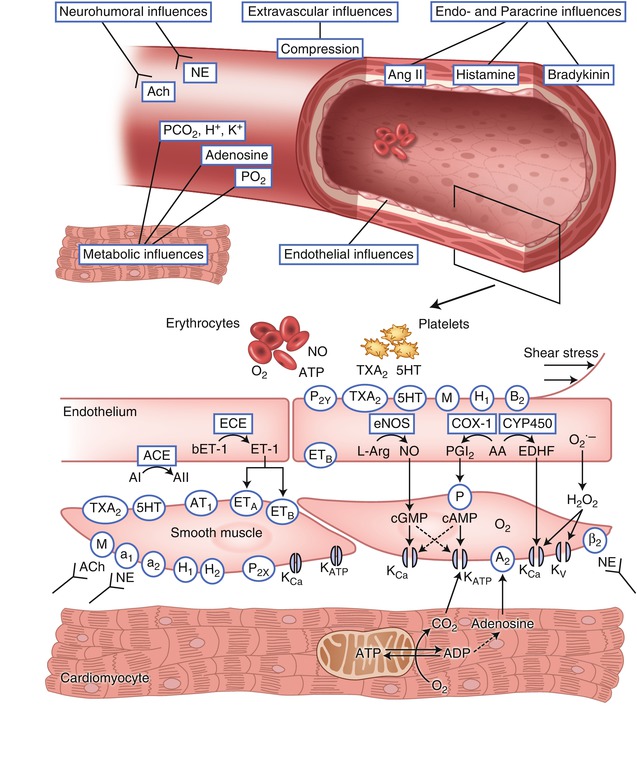
Fig. 4.1
Factors that determine vasomotor tone in coronary arterioles. ACh acetylcholine (receptor), NE norepinephrine, P 2X and P 2Y purinergic receptor subtypes X and Y that mediate ATP-induced vasoconstriction and vasodilation, respectively, TxA 2 thromboxane A2 (receptor), 5HT serotonin (receptor), M muscarinic receptor, H 1 and H 2 histamine receptors type 1 and 2, B 2 bradykinin type 2 receptor, ANG I and ANG II angiotensin I and II, AT 1 angiotensin receptor subtype 1, ET endothelin, ET A and ET B endothelin receptor subtypes A and B, A2 adenosine, β 2 β-adrenergic receptor, a 1 and a 2 α-adrenergic receptors, NO nitric oxide, eNOS endothelial NO synthase, PGI 2 prostacyclin, IP prostacyclin receptor, COX-1 cyclooxygenase, EDHF endothelium derived hyperpolarizing factor, CYP450 cytochrome P450 2C9, K Ca calcium sensitive potassium channel, K ATP ATP-sensitive potassium channel, K V voltage-sensitive potassium channel, AA arachidonic acid, L-Arg L-arginine. Receptors are indicated by an oval; enzymes are indicated by a rectangle. Normal endothelium produces factors that mediate vasodilation in response to stimulation of specific receptors by their agonists or increased shear force (Adapted from Duncker and Bache [1]; copyright American Physiological Society)
The downstream coupling of metabolic signals from the cardiac myocytes to the coronary resistance vessels involves potassium channels on the sarcolemma of the vascular smooth muscle cells [19]. Opening of these channels results in an outward flux of potassium, thereby increasing the membrane potential in the smooth muscle cell. This hyperpolarization causes voltage dependent calcium channels to close; the resultant decrease of intracellular calcium produces relaxation of the vascular smooth muscle and vasodilation [20]. Both ATP sensitive potassium channels (KATP) and calcium activated potassium channels (KCa) are abundantly expressed on coronary smooth muscle and contribute to regulation of blood flow. KATP channels act as metabolic sensors that open in response to decreases of intracellular ATP and/or increases of ADP. Pharmacologic blockade of KATP channel opening impairs coronary autoregulation, metabolic vasoregulation, and ischemic vasodilation [19]. Depletion of high energy phosphates (HEP) that occurs during ischemia would be expected to cause opening of KATP channels, but cannot account for coronary vasodilation during physiologic increases of myocardial O2 demands during exercise or other stress where bulk cytosolic HEP content is unperturbed. Rather, it is likely that ATP/ADP in a subsarcolemmal microenvironment (rather than bulk changes in cellular ATP) mediate regulation of KATP channel activity during physiologic conditions [21]. Although the factors that regulate vascular K+ ATP channel activity during physiologic conditions are not completely understood, the critical importance of this channel is demonstrated by the finding that mice with genetic deletion of coronary KATP channels are vulnerable to myocardial ischemia and sudden death [22]. KCa channels open in response to membrane depolarization and increased cytosolic Ca2+ concentrations, thereby providing negative feedback modulation of coronary vasoconstrictor responses [23]. KCa channels also respond to phosphorylation by cGMP and cAMP dependent protein kinases and are downstream effectors of vasodilators including adenosine, nitric oxide and β-adrenergic agonists. Thus, regulation of blood flow by the coronary resistance vessels involves a large number of redundant mechanisms which converge on sarcolemmal channels in the vascular smooth muscle that regulate membrane polarization and thus calcium flux and contractile activity of the smooth muscle (Table 4.2).
Table 4.2
Relative potency of vasodilators on coronary arteries and coronary arterioles in normal subjects (Normal) and in patients with coronary risk factors or atherosclerotic coronary disease and endothelial dysfunction (Dysfunction)
Coronary arteries | Coronary arterioles | |||
|---|---|---|---|---|
Normal | Dysfunction | Normal | Dysfunction | |
Acetylcholine | + + | Constrict | + | Constrict |
Nitrates | + + + + | + + + + | + | + |
Adenosine | + | ↔ | + + + + | + + + + |
Dipyridamole | + | ↔ | + + + | + + + |
Serotonin | Constrict | Constrict | + + + | + + + |
Nifedipine | + | ↔ | + + | + + |
Verapamil | + | ↔ | + | + |
Exercise | + | ↔ | + + + | + + + |
Myogenic Mechanisms
Myogenic automaticity refers to the intrinsic property of smooth muscle to respond to stretch with a counteracting increase in contractile force. Thus, an increase in intraluminal distending pressure causes the vascular smooth muscle to contract, while a decrease in pressure results in vasodilation. Myogenic activity is an intrinsic property of coronary arterioles and is not altered by endothelial denudation [1]. Myogenic responses are more prominent in arterioles from the subepicardium than from the subendocardium of the left ventricle, suggesting that intrinsic differences in responsiveness could contribute to the lesser ability to autoregulate in the subendocardium [24]. Although myogenic activity can be demonstrated in isolated coronary arterioles, the contribution of myogenic mechanisms to regulation of blood flow in the intact heart is likely of less importance than metabolic or endothelium-mediated responses.
Resistance Arteries
Small arteries (100–300 μm in diameter) contribute up to 40 % of total coronary resistance but, unlike the arterioles, these vessels are not responsive to the metabolic state of the myocardium [25]. However, when increased cardiac work results in metabolic vasodilation of the coronary arterioles, the resultant increase of blood flow causes shear-mediated endothelium-dependent dilation of the resistance arteries. Consequently, loss of flow mediated vasodilatation of the resistance arteries in patients in whom hyperlipidemia, atherosclerosis or hypertension has resulted in endothelial dysfunction can impair vasodilator reserve even in the absence of occlusive coronary artery disease. This is demonstrated by the finding that vasodilator reserve measured with positron emission tomography is impaired in patients with hyperlipidemia but with angiographically normal coronary arteries, and that this abnormality can be corrected by lipid lowering therapy [26].
Penetrating Arteries
Intramyocardial arteries can be divided into two classes; class A arteries arborize soon after entering the myocardium and supply principally the outer two-thirds of the left ventricular wall, while class B arteries penetrate deep into the myocardium before arborizing to supply blood to the subendocardium (Fig. 4.2) [27]. The class B vessels are also called penetrating arteries. The potential influence of the penetrating arteries on blood flow is documented by the finding that pressure in arterioles in the subendocardium is less than in subepicardial arterioles, indicating that a significant pressure loss occurs across the penetrating arteries [28]. Furthermore, vasomotor activity of the penetrating arteries can selectively influence blood flow to the subendocardium. Vasodilation of the penetrating arteries appears to be a mechanism by which nitrates augment blood flow to the subendocardium in ischemic myocardial regions [29].
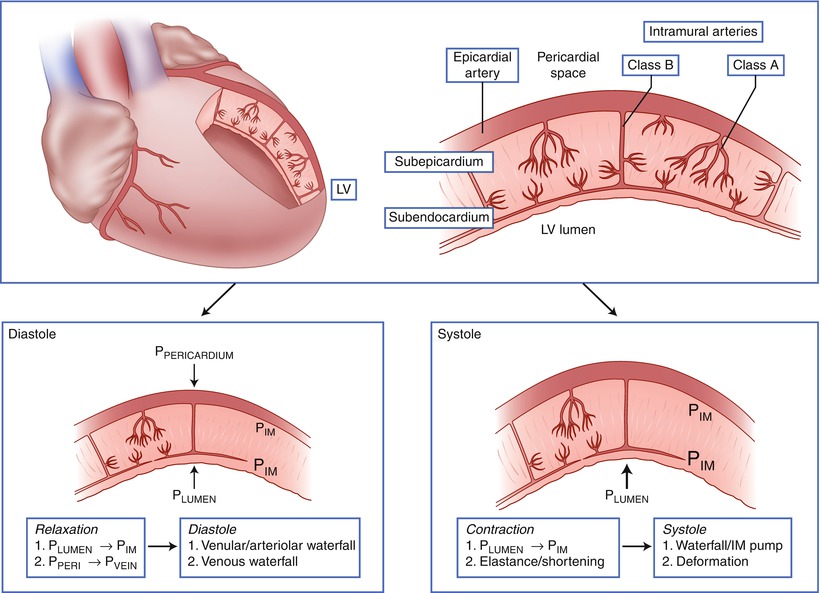
Fig. 4.2
Classification of intramural coronary vessels. Class A intramural arteries supply principally the outer two-thirds of the left ventricular wall, while Class B arteries penetrate deep into the myocardium before giving off resistance vessels and communicating with a vascular subendocardial plexus. During systole compressive forces from the contracting myocardium preferentially impede blood flow to the deeper myocardial layers. P IM intramyocardial pressure, P LUMEN pressure in the left ventricular lumen, P PERICARDIUM pressure in the pericardial space (Adapted from Duncker and Bache [1]; copyright American Physiological Society)
Endothelium-Dependent Vasodilation
Nitric Oxide
The importance of the vascular endothelium in mediating vasodilator responses was first demonstrated by Furchgott and Zawadski [30] who observed that acetylcholine caused relaxation of isolated coronary artery rings when the endothelium was intact, but produced contraction when the endothelium was removed. Subsequent studies demonstrated that a number of agonists including acetylcholine, bradykinin, histamine and thrombin cause coronary vasodilation indirectly by engaging specific receptors on the endothelium that trigger the release of one or more endothelium-derived relaxing factors (EDRF) (Fig. 4.1) [31]. The principal mediator of this vasodilation is nitric oxide (NO) which is produced when the constitutive endothelial cell enzyme nitric oxide synthase (eNOS) acts on arginine to produce citrulline with liberation of NO [32]. eNOS is a calcium-calmodulin dependent P450 enzyme; agonists that produce endothelium-dependent NO-mediated vasodilation generally act by causing increases of intracellular calcium to activate this enzyme. Alternatively, eNOS can be activated by serine phosphorylation, thereby causing sustained increases of NO production [33]. NO produced by eNOS in the endothelium can diffuse to the adjacent vascular smooth muscle and activate soluble guanylate cyclase; the resultant increase in cyclic guanosine monophosphate (cGMP) causes relaxation of the vascular smooth muscle. The shear force produced by blood flowing over the endothelial cells can also activate eNOS; in this way dilation of the epicardial arteries is coordinated with the increased flow produced by metabolic dilation of the coronary resistance vessels during exercise. eNOS is distinct from inducible nitric oxide synthase (iNOS), which is not present in normal coronary vessels, but can be induced by cytokines or inflammatory mediators, and which is not regulated by the intracellular calcium content.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


