Fig. 17.1
(a) The chemical structures for selected nitrate preparations. (b) Nitroglycerin’s physiologic effects in the heart and in the peripheral venous system. Nitroglycerin dilates large and medium-sized coronary arteries and improves myocardial blood flow to the subendocardial region. In the systemic circulation, nitroglycerin is a venodilator; therefore, it decreases venous return to the right heart and diminishes preload and wall tension, thereby decreasing myocardial oxygen demand. (c) The cellular biochemical effects of nitrates that correlate with their properties as coronary artery vasodilators. The nitrates increase guanylate cyclase activity, resulting in an increase in cyclic guanosine monophosphate (cGMP) production, which is associated with vasodilatation. It is believed that nitroglycerin exerts its endothelium-independent vasodilating effect through the activation of guanylate cyclase and the cellular increases in cGMP. GTP guanosine triphosphate
The physiologic effects of nitrates—increasing coronary blood flow and myocardial oxygen availability and decreasing myocardial oxygen demand—usually relieve angina promptly, that is, within 5–7 min. The commonly used nitrate preparations are listed in Table 17.1.
Table 17.1
Nitrate preparations used in the treatment of angina pectoris
Preparation | Dosage (mg) | Duration of effect | Frequency of administration |
|---|---|---|---|
Sublingual nitroglycerin | 0.3–0.5 | 15–30 min | For individual episodes |
Sublingual or chewable isosorbide dinitrate | 2.5–10 | 30–60 min | May be used instead of nitroglycerin |
Oral isosorbide dinitrate (Isordil) | 5–30 | 2 h | Every 2–3 h while patient is awake |
Oral isosorbide mononitrate (Ismo) | 10–20 | 24 h | Daily |
Oral isosorbide mononitrate (Imdur) | 30–60 | 24 h | Daily |
Oral isosorbide dinitrate (Isordil Tembids), longer-acting preparation | 40 | 6–8 h | Every 6–8 h |
Pentaerythritol tetranitrate (Peritrate) | |||
Oral | 10–40 | 3–4 h | Every 3–4 h |
Sustained | 80 | 8–10 h | Every 8–10 h |
Sustained-release oral nitroglycerin (Nitro-Bid) | 2.5–6.5 | 6 h | Every 6 h |
Nitroglycerin ointment | Thin film on 1–2 square inches of the anterior chest | 4–6 h | Every 4–6 h |
Nitroglycerin patches (sustained release) | 0.1, 0.2, 0.4 mg/h | Approximately 12 h | Every 12–24 h |
Nitroglycerin spray (Nitrolingual) | 1 puff prn | Few minutes | Prn for chest pain |
The various nitrate preparations differ primarily with regard to the time required for the onset of the antianginal effect, the duration of that effect, and the degree to which tolerance develops. The sublingual tablets and oral spray have the fastest onset of any of the preparations and are often used immediately before activity. Typically, relatively long-acting and orally administered nitrates, such as isosorbide dinitrate, are given at 6–8-h intervals, and isosorbide mononitrate is administered once or twice during the day [1, 4, 9]. In high-risk patients, a nitroglycerin patch or paste is applied during the nighttime hours and removed the next morning after the patient arises. This approach maximizes the systemic availability of the nitrate and reduces the development of tolerance to the drug [10, 11]. Patients with stable angina should be cautioned about the absolute contraindication to the concomitant use (within 24 h of one another) of nitrates, sildenafil, and other similar erectile dysfunction drugs that increase local concentration of cGMP (i.e., vardenafil and tadalafil). This combination may lead to serious, prolonged, and life-threatening hypotension [12].
β-Adrenergic Antagonists
β-adrenergic antagonists attenuate heart rate, systolic blood pressure, and contractile responses at rest and during exercise (Tables 17.2, 17.3, and 17.4). Through these effects, selected beta-blockers have been shown in numerous trials to have mortality benefits in patients with hypertension, previous myocardial infarction (MI), and heart failure [13, 14]. Beyond mortality benefits, reductions in heart rate-systolic blood pressure for any particular level of activity may reduce myocardial oxygen demand enough to allow a patient to engage in a particular activity without angina, whereas previously that was not possible. This is primarily accomplished through heart-rate reductions that preferentially prolong diastole and, therefore, the time during which coronary perfusion occurs. The beta-blockers most commonly used in the treatment of stable angina are listed in Table 17.5. Beta-blockers are classified as β1– or nonspecific beta-blockers (Fig. 17.2); see also (Table 17.5) [15–22]. β1-specific blockers, such as metoprolol, alter heart rate and myocardial contractile responses but, at low doses, may interfere less with smooth muscle dilatation. At higher doses, the “selective” beta-blockers have physiologic effects more like those of nonspecific beta-blockers and may attenuate bronchial and smooth muscle dilatation and exacerbate bronchospasm. Nonspecific beta-blockers, such as propranolol, reduce heart rate and myocardial contractile state and interfere with bronchial and vascular smooth muscle dilatation. Therefore, the β1-specific blockers given in reduced dosage may have certain advantages in patients with chronic obstructive pulmonary diseases. They may also reduce insulin release less than nonspecific blockers and therefore may be advantageous in the treatment of selected patients with diabetes.
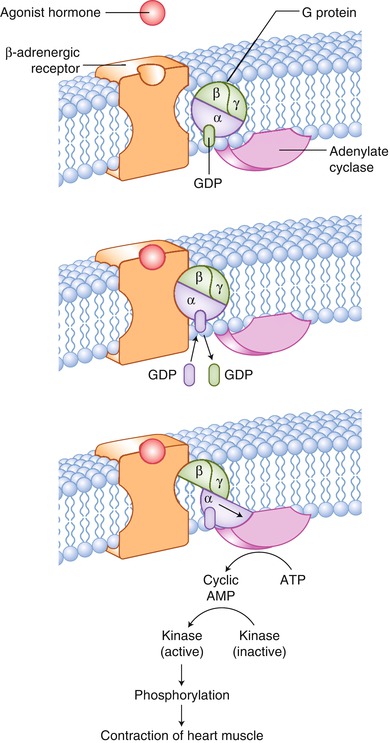
Table 17.2
Side effects of β-adrenergic antagonists
Easy fatigability |
Insomnia |
Dizziness or syncope |
Dyspnea with effort |
Sexual impotence |
Bronchospasm |
Bradycardia |
Heart block |
Hypotension |
More difficult to recognize hypoglycemia in insulin-dependent diabetic patients |
Table 17.3
Indications for β-adrenergic antagonists in patients with stable angina
Prevent development of angina at relatively low heart rate–systolic blood pressure product |
Treat exercise-induced ventricular arrhythmias |
Treat systemic arterial hypertension |
Table 17.4
Relative contraindications to the administration of β-adrenergic antagonists
Severe congestive heart failurea | Severe peripheral vascular disease |
Marked bradycardia (heart rate <55 beats/min) | Insulin-dependent diabetes mellitus that is poorly controlled and labile |
Advanced atrioventricular block (first-, second-, or third-degree) | Sexual impotence |
Bronchospasm | |
Systemic arterial hypotension (systolic blood pressure <90 mmHg) | Coronary artery spasm |
Table 17.5
Selected β-adrenergic antagonists
Name | Beta-blockade potency ratio (propranolol = 1.0) | Cardioselective | Usual therapeutic dose range (mg/day) | Elimination half-life (h) | Route of excretion |
|---|---|---|---|---|---|
Propranolol (nonspecific) | 1.0 | 0 | 80–480 | 3.5–6.0 | Urine |
Timolol | 6.0 | 0 | 5–40 | 4–5 | Urine |
Oxprenolol | 0.5–1.0 | 0 | 40–360 | 2 | Urine |
Sotalol | 0.3 | 0 | 80–480 | 5–13 | Urine |
Metoprolol (β1) | 1.0 | + | Given IV at 5 mg in each of 3 doses at 2-min intervals, given orally at 100–800 | 3–4 | Urine |
Pindolol | 6.0 | 0 | 2.5–30.0 | 3–4 | Urine |
Atenolol (β1) | 1.0 | + | 100–400 | 6–9 | Approximately 40 % of unchanged drug in urine |
Alprenolol | 0.3 | 0 | 200–800 | 2–3 | Urine |
Acebutolol (β1) | 0.3 | + | 400–800 | 8 | Uncertain |
Nadolol | 0.5–1.0 | 0 | 40–80 | 20–24 | Urine |
Sotalola | 1.0 | 1 | 80–640 | 12 | Urine |
Esmololb (β1) | 0.025 | + | Given IV at 25–300 mg/kg/min | 12 | Hydrolysis by plasma |
Labetalol | 0.3 | Selective alpha- and nonselective beta-blockers | Given IV 0.25 mg/kg over 2 min and additional 40–80 mg at 1-h intervals, given orally at 200–400 | 8–10 | Urine and bile |
Toprol (β1) | Selective long-acting metoprolol | 50–100 | 24 | ||
Carvedilol | Selective alpha- and nonselective beta-blockers | 6.25–50 | 7–10 |
Fig. 17.2
The cellular basis for the ability of β-adrenergic antagonists to interfere with agonist stimulation of β-adrenergic receptors. Released from synaptic terminals, catecholamines enhance cardiac output and maintain arterial diffusion pressure. β-adrenergic receptors bind the catecholamines. Catecholamines are released at the synapse, leading to enhanced heart rate and contractile force, through the following mechanisms: (1) sympathetic nerve terminals release norepinephrine, which binds to the β-adrenergic receptor, activating adenylate cyclase through the coupling effect of G proteins; (2) the increase in intracellular cyclic adenosine monophosphate (AMP) leads to the activation of protein kinase A; and (3) protein kinase A phosphorylates a variety of proteins, which enhances their catalytic activity, promoting a calcium-dependent increase in cardiac contractility. ATP adenosine triphosphate, GDP guanosine diphosphate, GTP guanosine triphosphate
Beta-blockers may cause bradycardia, bronchospasm, hypotension, atrioventricular (AV) block, and depression of myocardial contractility. They may also exacerbate coronary artery spasm and make it more frequent and severe. Therefore, they should not be used in patients with bradycardia, hypotension, AV block, severe bronchopulmonary lung disease (especially in those with bronchospasm), or coronary artery spasm. Beta-blockers are used with great caution, and initially in small doses, when they are used in patients with clinically severe heart failure. They should also be used with caution in patients with important peripheral vascular disease and insulin-dependent diabetes mellitus, particularly when the blood glucose level has been labile and difficult to control. Increasing in popularity are sustained-release beta-blockers that can be administered once a day and have a sustained release into the systemic circulation, resulting in the attenuation of β-adrenergic responses throughout the day (Table 17.5). A beta-blocker should be considered as therapy in patients who experience angina at a relatively low level of physical activity or stress. Beta-blockers are often used in conjunction with nitrates to treat exercise- or stress-related angina.
Calcium Antagonists
Slow calcium channel antagonists alter slow calcium channel transport into the cell (Fig. 17.3 and Tables 17.6, 17.7, and 17.8) [23–46]. As a result, these agents relax vascular smooth muscle and increase coronary blood flow. The slow calcium channel antagonists are divided into two major classes: dihydropyridines and nondihydropyridines (which comprise the phenylalkylamines and modified benzothiazepines).
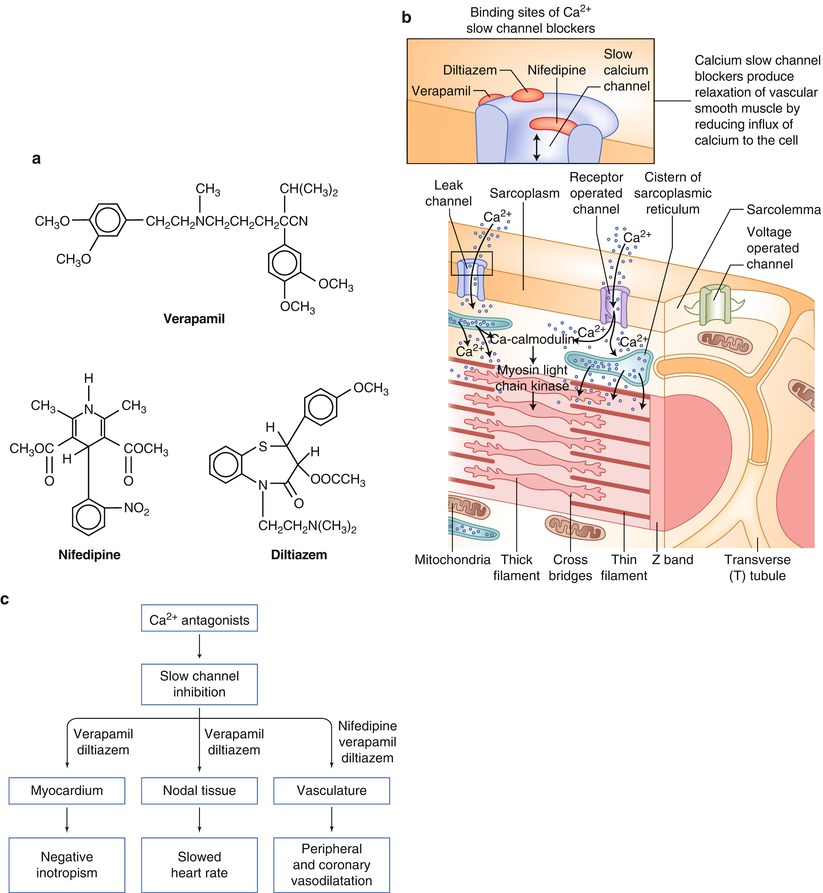
Fig. 17.3
(a) The chemical structures for selected slow calcium channel antagonists currently available. (b) The cellular sites of action for the slow calcium channel antagonists. (c) The location and type of effect produced by each of the slow calcium channel antagonists
Table 17.6
Selected prototype slow calcium channel blockers
Dosage | Onset of action | Therapeutic plasma concentration | Metabolism | Excretion | |||
|---|---|---|---|---|---|---|---|
Oral | IV | Oral | IV | ||||
Diltiazem | 30–90 μg | 75–150 μg/kg | >30 min | >10 min | 50–200 ng/mL | Deacetylation | 60 % fecal |
(10–20 mg) | N-Demethylation | ||||||
O-Demethylation | |||||||
Nifedipine (dihydropyridine calcium antagonist) | 10–40 mg | 5–15 μg/kg | >20 min | >5 min | 25–100 ng/mL | Alpha-hydroxycarboxylic acid and alpha-lactone with no known activity | 20–40 % fecal |
q 6–8 h | (3 min SL) | 50–80 % renal | |||||
Verapamil | 80–120 mg | 150 μg/kg | >30 min | >5 min | <100 ng/mL | N-dealkylation | 15 % fecal |
q 6–12 h | (10–20 mg) | N-demethylation | 70 % renal | ||||
Major hepatic first-pass effect | |||||||
Table 17.7
Selected newer slow channel calcium antagonists
Type | Dosage | Special uses | Excretion |
|---|---|---|---|
Nisoldipine (dihydropyridine) | 10 mg PO q 12 h | Treat systemic arterial hypertension and angina | Renal |
Felodipine (dihydropyridine) | 5–10 mg PO q 24 h | Treat systemic arterial hypertension and angina | Renal |
Nicardipine (dihydropyridine) | 20–40 mg PO t.i.d. | Treat systemic arterial hypertension and angina | Renal |
Nimodipine (dihydropyridine) | 60 mg q 4 h PO for 21 days | Reduce cerebral vascular ischemia after subarachnoid hemorrhage | Renal |
Isradipine (dihydropyridine) | 2.5 mg PO b.i.d. increasing up to 20 mg PO qd | Hypertension | Renal |
Amlodipine (dihydropyridine) | 5–10 mg PO qd | Systemic arterial hypertension and angina | Bile and renal |
Bepridil (Na+ and Ca2+ channel blocker) | 200 mg PO qd increasing to 400 mg PO qd | Angina | Renal and fecal |
Table 17.8
Clinical indications for administration of slow calcium channel antagonists to patients with stable angina
Chest pain at a relatively low level of exercise or stress; in these cases, a calcium antagonist can be used alone or in combination with nitrates, a beta-blocker, or botha |
Systemic arterial hypertension |
Atrial arrhythmiasb |
Exercise-induced ventricular tachycardiac |
The nondihydropyridines are distinguished from the dihydropyridines primarily by their effect on AV node conduction. Two of these slow calcium channel antagonists, verapamil and diltiazem, slow the heart rate by decreasing sinus node impulse formation and AV conduction. Therefore, verapamil and diltiazem have some of the same hemodynamic effects as beta-blockers in that they reduce myocardial oxygen demand at rest and during exercise by attenuating heart rate and contractile responses. However, they also increase coronary blood flow, primarily to epicardial regions supplied by severely narrowed coronary arteries.
Nifedipine, a dihydropyridine calcium antagonist, does not decrease impulse formation in the sinus node or delay AV conduction. Therefore, it does not decrease heart rate but may actually increase it. The dihydropyridine calcium antagonists are potent vasodilators, causing coronary artery vasodilatation. Nifedipine, as the prototypical dihydropyridine calcium antagonist, dilates coronary arteries, increasing blood flow to the epicardial regions supplied by significantly narrowed coronary arteries. Amlodipine, a second-generation dihydropyridine, has greater antihypertensive effects and may reduce cardiovascular events independent of these effects [47].
In addition to their negative chronotropic effects, the nondihydropyridines also show significant negative inotropic effects. Verapamil has a marked negative inotropic effect on the heart and should not be given to patients with clinically important congestive heart failure (CHF). Diltiazem has a lesser negative inotropic effect, but it should be given very carefully to patients with CHF. Great care should be used in combining diltiazem with another negative inotropic agent, such as a beta-blocker, in patients with clinically important CHF. Nifedipine may be given with relative safety to patients with important CHF. Its negative inotropic effect is masked by its ability to reduce systemic vascular resistance, which enables the heart to contract more effectively against a reduced afterload. As noted previously, beta-blockers are used in the treatment of selected patients with severe CHF; doses are very small initially and are slowly increased.
Each of the slow calcium channel antagonists has important side effects (Table 17.9). With nifedipine administration, many patients describe a flushing sensation, dizziness, and palpitations, which result from its systemic vasodilating effect. Peripheral edema occurs in patients who receive nifedipine (and other dihydropyridine calcium antagonists) and is probably the result of venodilatation. Regarding verapamil, constipation is its most common side effect, although symptoms related to CHF, bradycardia, or advanced AV block may also occur. The combination of verapamil with a beta-blocker is particularly potent in reducing heart rate, systemic blood pressure, and contractile state. It should be used with extreme caution in patients with CHF, AV block, hypotension, or bradycardia. Diltiazem is usually the best tolerated of the slow calcium channel antagonists. When side effects occur, they are usually related to bradycardia or increasing CHF. A nondihydropyridine calcium antagonist can be used as an alternative to a beta-blocker for treating patients with stable angina. However, there has been considerable concern about the potential adverse effects of the calcium antagonists in patients with coronary heart disease (CHD)—especially those with acute coronary syndromes (ACS), such as unstable angina or acute myocardial infarction (AMI)—when relatively short-acting dihydropyridine calcium antagonists, like nifedipine, are given in doses of 80 mg or more per day. Some data suggest an increase in mortality and a lack of clinical benefit when this agent is given in larger doses to patients with ACS [48].
Table 17.9
Side effects of slow calcium channel antagonists
Verapamil or diltiazem | Nifedipine |
|---|---|
Marked bradycardia | Flushing sensation |
Hypotension | Dizziness |
Constipation | Hypotension |
Congestive heart failure | Peripheral edema |
Skin rash | Tachycardia |
Heart block | Skin rash |
A calcium antagonist can be combined with nitrates and a beta-blocker to treat patients who experience angina at low levels of effort. This combination of pharmacologic agents may be useful in enabling patients to be more active without having angina. Clinically, the safest combination of a beta-blocker with a calcium antagonist is to use a dihydropyridine calcium antagonist, such as nifedipine. The next safest clinical combination is a beta-blocker and diltiazem. One should initiate combined therapy with a beta-blocker and diltiazem or verapamil by using relatively small doses of the calcium antagonist and gradually increasing them when the patient’s hemodynamic and clinical responses are consistent with the safety of the combined regimen.
Angiotensin-Converting Enzyme Inhibitors
Angiotensin-converting enzyme inhibitors, unlike previously mentioned nitrates, beta-blockers, and calcium channel antagonists, do not diminish the symptoms of stable angina. Nonetheless, growing evidence indicates that they provide significant benefit in specific groups of patients with stable angina. Also, unlike beta-blockers and calcium antagonists, the benefits of ACE inhibitors are probably independent of their effects on blood pressure. For example, the Trial on Reversing Endothelial Dysfunction (TREND) investigators showed that ACE inhibition with quinapril improved endothelial dysfunction in patients who were normotensive and who did not have severe hyperlipidemia or heart failure [49].
The Heart Outcomes Prevention Evaluation (HOPE) was a large-scale, multicenter (267 hospitals in 19 countries), randomized, placebo-controlled trial of ACE inhibitor therapy and vitamin E supplementation in patients at high risk for vascular events [50]. The inclusion criteria included age greater than 55 years, evidence of vascular disease (CHD, stroke, or peripheral vascular disease) or diabetes, and one other cardiovascular risk factor. Patients with heart failure, a low left ventricular ejection fraction, current ACE inhibitor or vitamin E therapy, or acute events within the previous 4 weeks were excluded. A total of 9,541 qualifying patients received ramipril (up to 10 mg/day), placebo and vitamin E (400 IU/day), or placebo only and were followed for 4–6 years. The primary end point of the study was the composite of cardiovascular death, MI, or stroke. Secondary end points included revascularization and the development of CHF, unstable angina, or complications of diabetes.
Although vitamin E therapy was not associated with any significant clinical benefit, ramipril therapy was associated with a highly significant clinical benefit (Fig. 17.4). Composite primary outcome events occurred in 13.9 % of the ramipril group and 17.5 % of the placebo-treated group [risk ratio (RR), 0.78; p = .000002]. There was also significant benefit in the individual endpoints of cardiovascular death (6.0 % vs. 8.0 %; RR, 0.75; p = .002), MI (9.8 % vs. 12.0 %; RR, 0.80; p = .0005), stroke (3.3 % vs. 4.8 %; RR, 0.68; p = .0002), and total mortality (10.3 % vs. 12.2 %; RR, 0.83, p = .035). There were no differences between groups in noncardiovascular death (4 % vs. 4 %; p = NS). Regarding the secondary end points, ramipril therapy had no effect on the development of unstable angina, but there was a trend toward fewer heart failure hospitalizations, and there were significantly fewer revascularizations (16.0 % vs. 18 %; RR, 0.85; p = .0013).
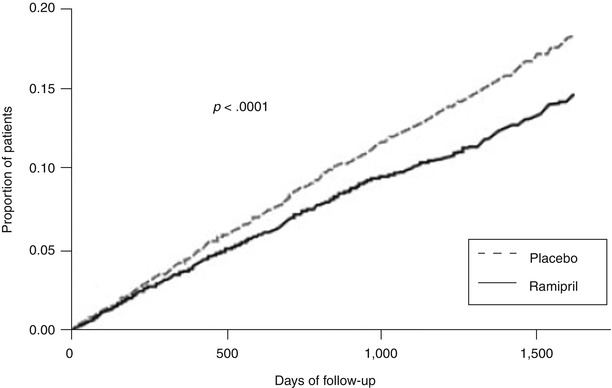
Fig. 17.4
Kaplan-Meier estimates of the composite outcome of myocardial infarction, stroke, or death from cardiovascular causes in the ramipril group and the placebo group. The relative risk of the composite outcome in the ramipril group was 0.78 (95 % confidence interval, 0.70–0.86) (From Yusuf et al. [50]. Reprinted with permission from Massachusetts Medical Society)
Ramipril also had a striking benefit in patients with documented normal left ventricular ejection fractions (n = 4,676; mean ejection fraction, 59 %): the drug significantly reduced the frequencies of the primary outcome end point (13.6 % vs. 18.3 %), cardiovascular death (5.0 % vs. 7.0 %), MI (10.3 % vs. 13.5 %), stroke (2.9 vs. 4.2 %), heart failure (8.3 % vs. 10.4 %), and revascularization (19.8 % vs. 23.8 %) (Table 17.10). In the overall cohort, the divergence of the primary end point was present in the first year, and the event curves continued to diverge through year 4. The clinical benefit was noted in virtually all of the major subgroups, including patients with or without cerebrovascular disease, diabetes, hypertension, coronary artery disease (CAD), or peripheral vascular disease, and in men or women and in young or old.
Table 17.10
Incidence of the primary outcome and of death from any cause in the HOPE trial
Outcome | Ramipril group n = 4,645 (%) | Placebo group n = 4,652 (%) | Relative risk 95 % CI (%) | z statistic | p valuea |
|---|---|---|---|---|---|
Myocardial infarction, stroke, or death from cardiovascular causesb | 651 (14.0) | 826 (17.8) | 0.78 (0.70–0.86) | −4.87 | <0.001 |
Death from cardiovascular causesc | 282 (6.1) | 377 (8.1) | 0.74 (0.64–0.87) | −3.78 | <0.001 |
Myocardial infarctionc | 459 (9.9) | 570 (12.3) | 0.80 (0.70–0.90) | −3.63 | <0.001 |
Strokec | 156 (3.4) | 226 (4.9) | 0.68 (0.56–0.84) | −3.69 | <0.001 |
Death from noncardiovascular causes | 200 (4.3) | 192 (4.1) | 1.03 (0.85–1.26) | 0.33 | 0.74 |
Death from any cause | 482 (10.4) | 569 (12.2) | 0.84 (0.75–0.95) | −2.79 | 0.005 |
Mechanistically, there was only a minor decrease in systolic (−2.17 mmHg) and diastolic (−3.13 mmHg) blood pressure, and the investigators speculated that although there was a strong relationship between clinical events and systolic blood pressure (but not diastolic blood pressure), the primary benefits of ramipril therapy were vascular rather than a blood pressure-lowering effect. In this population, the number of patients treated (with 4 years of therapy) needed to prevent one primary clinical event was 6; treating 1,000 patients would prevent 170 adverse clinical events.
The HOPE study provides strong evidence that in patients at risk for vascular events, ACE inhibitor therapy with ramipril significantly reduces cardiovascular death, MI, stroke, heart failure, and revascularization (Tables 17.10 and 17.11), even in patients with documented normal left ventricular ejection fractions. Vitamin E therapy did not appear to be beneficial.
Table 17.11
Incidence of secondary and other outcomes in the HOPE trial
Ramipril group n = 4,645 (%) | Placebo group n = 4,652 (%) | Relative risk (95 % CI) | z statistic | p valuea | |
|---|---|---|---|---|---|
Secondary outcomesb | |||||
Revascularization | 742 (16.0) | 852 (18.3) | 0.85 (0.77–0.94) | −3.17 | 0.002 |
Hospitalization for unstable angina | 554 (11.9) | 565 (12.1) | 0.98 (0.87–1.10) | −0.41 | 0.68 |
Complications related to diabetesc,d | 299 (6.4) | 354 (7.6) | 0.84 (0.72–0.98) | −2.16 | 0.03 |
Hospitalization for heart failure | 141 (3.0) | 160 (3.4) | 0.88 (0.70–1.10) | −1.16 | 0.25 |
Other outcomes | |||||
Heart failurec | 417 (9.0) | 535 (11.5) | 0.77 (0.67–0.87) | −4.09 | <0.001 |
Cardiac arrest | 37 (0.8) | 59 (1.3) | 0.62 (0.41–0.94) | −2.28 | 0.02 |
Worsening anginac | 1,107 (23.8) | 1,220 (26.2) | 0.89 (0.82–0.96) | −2.91 | 0.004 |
New diagnosis of diabetese | 102 (3.6) | 155 (5.4) | 0.66 (0.51–0.85) | −3.31 | <0.001 |
Unstable angina with electrocardiographic changesb | 175 (3.8) | 180 (3.9) | 0.97 (0.79–1.19) | −0.30 | 0.76 |
The European Trial on Reduction of Cardiac Events with Perindopril in Stable Coronary Artery Disease (EUROPA) was a study of 13,655 patients with stable CAD but with no apparent heart failure, including 64 % with a previous MI, 61 % with angiographic evidence of CAD, 55 % with prior coronary revascularization, and 5 % with only a positive stress test [51]. The primary end point was cardiovascular death, MI, or cardiac arrest, determined over a mean follow-up of 4.2 years. Primary end-point events occurred in 10 % of placebo-treated patients and 8 % of perindopril-treated patients (p = 0.003). The number needed to treat (NNT) to prevent one event over 4 years was approximately 50 patients. Combined with the HOPE trial, this study strengthened the role of ACE inhibition in patients with CAD.
The Prevention of Events with Angiotensin Converting Enzyme Inhibition (PEACE) study was designed to assess the efficacy of ACE inhibitor therapy in patients with stable CAD and normal or slightly reduced left ventricular function [52]. A total of 8,290 patients were included in this double-blind, randomized, placebo-controlled trial, which compared trandolapril (n = 4,158, target dose 4 mg/day) to placebo (n = 4,132). Baseline therapy included lipid-lowering therapy in 70 % and prior coronary revascularization in 72 %. The incidence of the primary end point (the composite of cardiovascular death, MI, or revascularization) was 21.9 % in the trandolapril group and 22.5 % in the placebo group (p = NS) over a median follow-up of 4.8 years. Thus, PEACE showed that the benefits of ACE inhibitors noted in studies of higher-risk patients, such as HOPE and EUROPA, do not necessarily extend to a lower-risk population. Of note, the event rate in the placebo arm of PEACE was lower than that in the ACE inhibitor arms of HOPE and EUROPA. The ACE inhibitors clearly play an important role in the care of some patients with stable angina. Defining the exact extent of that role and the exact subpopulation that can most benefit remains a task for the future.
Platelet Antagonists
When endothelial injury occurs, platelets aggregate after attaching to the subendothelial collagen and other matrix proteins exposed by the endothelial injury. Platelet aggregation may mechanically obstruct severely narrowed coronary arteries and is associated with the accumulation of mediators that promote further platelet aggregation and dynamic vasoconstriction, including thromboxane A2, serotonin, thrombin, platelet-activating factor, ADP, oxygen-derived free radicals, tissue factor, and endothelin [53–59].
Aspirin
In patients at increased risk for MI with known or suspected CAD, this risk may be reduced by the administration of aspirin [60–63]. Aspirin is an inhibitor of platelet and endothelial cyclooxygenase (COX-1 and −2) and thus reduces platelet thromboxane and endothelial cell prostacyclin formation (Fig. 17.5). Its effect on platelet COX is irreversible and persists for the lifetime of exposed platelets—approximately 11 days. With initial therapy, higher doses of aspirin are required to decrease endothelial cell COX activity; therefore, low-dose aspirin tends to reduce thromboxane levels more than prostacyclin concentration, but long-term administration of aspirin, even in a low dose, may reduce prostacyclin concentrations as well. Inhibiting the release of thromboxane A2 also attenuates platelet aggregation in vivo. Aspirin’s weaker effect, reducing COX-2 activity, may potentially reduce inflammation, and at least some of its beneficial effects may be the result of its decreasing the vulnerability of unstable atherosclerotic plaques by its anti-inflammatory effects.
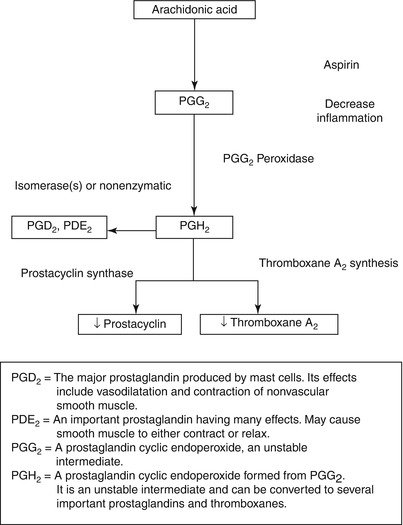
Fig. 17.5
The scheme for the synthesis of thromboxane A2 (TXA2) and prostacyclin (PGI2) from arachidonic acid in platelets and endothelial cells. Aspirin’s inhibitory effect is at the cyclooxygenase step, where it inhibits this enzyme and thereby diminishes the synthesis of both TXA2 and prostacyclin. TXA2 synthesis inhibitors interfere with the conversion of PGH2 to TXA2 through TXA2 synthase. TXA2 receptor antagonists simply antagonize the effects of TXA2 on platelets and vascular tissue
Much of the support for the routine use of aspirin in secondary prevention comes from the Antithrombotic Trialists’ meta-analyses. The first meta-analysis (the Antiplatelet Trialists Collaboration), which was published in 1994 and included clinical trials up through 1990, showed that oral antiplatelet therapy (primary aspirin) was effective in preventing recurrent events across a wide range of atherosclerotic vascular disease, primarily coronary and cerebrovascular [64–66]. A more comprehensive analysis, which involved trials up through 1997 and which placed additional focus on stroke and peripheral arterial disease, was published in 2002 [67]. The results showed that overall, antiplatelet therapy (primarily aspirin) reduced the incidence of any serious vascular event by one quarter, nonfatal MI by one third, nonfatal stroke by one quarter, and vascular mortality by one sixth (Fig. 17.6). The absolute reduction in serious vascular events was 36 per 1,000 patients treated for 2 years after stroke or transient ischemic attack (TIA). In 21 trials of patients with stroke or TIA, antiplatelet therapy reduced vascular events from 21.4 % (with control treatment) to 17.8 %.
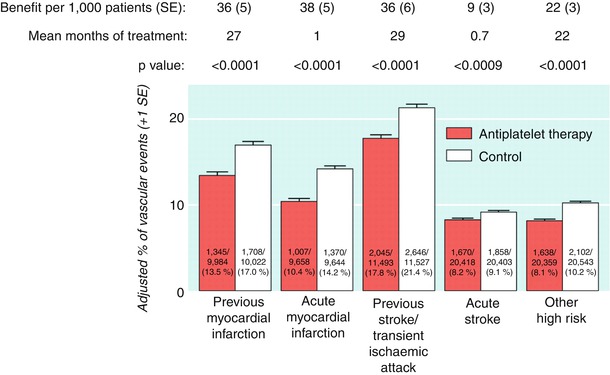
Fig. 17.6
Absolute effects of antiplatelet therapy (clopidogrel + aspirin) on vascular events (myocardial infarction, stroke, or vascular death) in the five main high risk categories in the Antithrombotic Trialists’ Collaboration’s meta-analysis of data from randomized, controlled trials. Adjusted control totals were calculated after any unevenly randomized trials were converted to even ones by counting control groups more than once. (From the Antithrombotic Trialists’ Collaboration [67]. Reprinted with permission from the BMJ Publishing Group Limited)
The amount of aspirin required to protect patients is not well established. In the meta-analysis described above, aspirin doses of 75–150 mg daily were at least as effective as higher doses. The effect of doses less than 75 mg is less certain. There was no good evidence to support the hypothesis that doses of aspirin ≥1,000 mg daily might be preferable in patients at high risk of stroke. Furthermore, a more recent trial (not included in the meta-analysis)—the Aspirin and Carotid Endarterectomy Trial—showed that in patients undergoing carotid endarterectomy, the composite outcome of MI stroke or death was significantly lower among patients taking 81 or 325 mg of aspirin than in those taking 625–1,300 mg [68].
The Harvard Physicians’ Study suggested that one aspirin every other day reduced the risk for MI in male physicians believed to be at increased risk. However, a British study in which one aspirin per day was administered failed to show protection against the development of MI [61, 63]. Administering aspirin to patients after MI reduces the risk of recurrent infarction and death, especially in patients with non-Q-wave infarcts [60]. Although the optimal protective dose of aspirin in patients with CAD is not known, many physicians recommend administering one 325-mg aspirin every other day to one aspirin every day in individuals believed to be at risk for future coronary events. The long-term administration of aspirin, even at a low dose, may decrease vascular prostacyclin concentration. Theoretically, this may be disadvantageous over time, because prostacyclin is an endogenous endothelial vasodilator and an inhibitor of platelet aggregation. However, no adverse clinical consequence has been reported.
The authors recommend one aspirin (81–325 mg) every other day to one aspirin every day in patients believed to be at increased risk for future coronary events and in whom there is no contraindication. Administration of aspirin with consequent COX inhibition and reduction in prostacyclin concentration may be associated with a reduction in renal blood flow and a rise in serum blood urea nitrogen and creatinine levels. Nonsteroidal antiinflammatory agents that are COX inhibitors may also cause a reduction of renal blood flow and a decline in renal function. Periodic measurements of blood urea nitrogen and creatinine concentrations are advised once aspirin therapy is begun. There is a risk for gastritis and gastrointestinal ulceration and bleeding when aspirin is administered, and some patients develop asthma (Table 17.12). Therefore, long-term aspirin therapy necessitates careful patient selection and follow-up.
Table 17.12
Potential side effects of aspirin
Gastritis |
Stomach or gastrointestinal ulceration |
Gastrointestinal bleeding |
Easy bruising and bleeding with minor trauma |
Asthma |
Decline in renal function |
Thrombocytopenia |
Thienopyridines
Another class of antiplatelet agents that may be beneficial in patients with stable angina are the thienopyridines. This class of drugs includes clopidogrel (the most widely used thienopyridine), ticlopidine, prasugrel, and ticagrelor.
Thienopyridines act to inhibit the platelet 2-methylthio-adenosine diphosphate (ADP)-binding receptor [69–76]. In response to other stimuli whose actions may be mediated in part through ADP released from endogenous platelet granules, thienopyridines also blunt platelet aggregation. Thienopyridines also inhibit platelet aggregation in response to shear stress, and they deaggregate platelet thrombus that has already formed. It is generally believed that thienopyridines are biotransformed and activated in the liver; biotransformation of the thienopyridines by the hepatic CP450 system has been well documented, and plasma levels of the parent drug are not detectable 2 h after oral administration. It has been suggested that both ticlopidine and clopidogrel can interfere with in vitro ADP-induced aggregation of washed human platelets; thus, biotransformation may not be a necessary step [76]. This effect was not noted when either plasma or albumin was present. Regardless of the exact mechanism, thienopyridines produce a permanent inhibition of the low-affinity ADP receptor, and platelets exposed to thienopyridines are irreversibly inhibited for their lifetime (8–10 days).
The onset of action of ticlopidine is about 48–72 h, and it takes about 5–6 days to achieve steady-state levels of platelet inhibition. Ticlopidine has also been shown to reduce fibrinogen concentrations and blood viscosity, and it increases the filterability of whole blood and red blood cells. Ticlopidine is metabolized in the liver. Adverse effects of ticlopidine include neutropenia, rash, diarrhea, and, rarely, thrombotic thrombocytopenic purpura [71]. For these reasons, ticlopidine is no longer widely used.
Clopidogrel bisulfate is a thienopyridine derivative. The antiplatelet effects of clopidogrel have a somewhat more rapid onset than those of ticlopidine, particularly with oral loading. Clopidogrel is rapidly absorbed after oral administration, reaching peak plasma concentrations approximately 1 h after an oral dose. Steady-state concentrations are reached after approximately 3 days of consecutive dosing. Food and antacids do not appear to interfere with its absorption or bioavailability. Both clopidogrel and its primary metabolite (a carboxylic acid derivative) are highly protein bound (in vitro binding to albumin of 98 and 94 %, respectively). The drug is well absorbed in the elderly and has comparable pharmacodynamic effects in elderly and younger patients. Evaluation of the pharmacodynamic effects of clopidogrel has shown significant inhibition of platelet function within 2 h of a 400 mg dose, an effect that persists for up to 48 h. With repeated oral daily doses of 50–100 mg, a significant antiplatelet effect is measurable at 48 h and reaches a steady state 4–7 days after therapy begins. Dose ranging studies have shown that a clopidogrel dose of 75 mg/day provides a similar degree of platelet inhibition to that achieved with 250 mg b.i.d. of ticlopidine. The side effects of clopidogrel are the same as or even milder than those of aspirin and are much less frequent than those of ticlopidine. The much lower frequency of thrombotic thrombocytopenic purpura and bone marrow toxicity, even over an extended period of follow-up, is a major advantage of clopidogrel over ticlopidine [69].
No major trials have yet been completed on the efficacy of ticlopidine in patients with stable angina. The effect of this drug must therefore be extrapolated from the results of trials conducted in stroke patients with risk factors similar to those of patients with stable angina. Two large studies have examined the utility of ticlopidine in patients with cerebrovascular disease. The Canadian American Ticlopidine Study (CATS) trial compared ticlopidine with placebo in patients with a history of recent stroke [77]. The primary end point was the composite of ischemic stroke, MI, or vascular death. The placebo group had a primary event greater than the ticlopidine group [relative risk reduction (RRR), 23.3 %; p = .02]. The Ticlopidine Aspirin Stroke Study (TASS) trial compared ticlopidine with aspirin in patients with a history of a recent stroke precursor or minor stroke within the past 3 months [78]. The primary end point in this trial was the composite of nonfatal stroke and all-cause mortality. The primary event rate was higher in the aspirin group (19 %) than the ticlopidine group (17 %) (RRR, 12 %; p = .048).
The Clopidogrel versus Aspirin in Patients at Risk of Ischemic Events (CAPRIE) trial was a large-scale randomized trial of the safety and efficacy of clopidogrel (75 mg/day) versus aspirin (325 mg/day) in 19,185 patients with atherosclerotic vascular disease who were followed for up to 3 years [79]. The study cohort included patients with recent ischemic stroke (within 6 months), recent MI (within 35 days), or symptomatic peripheral arterial disease. The primary end point was the composite incidence of stroke (fatal and nonfatal), MI (fatal and nonfatal), and other vascular death. At a mean follow-up of 1.9 years, the clopidogrel group had significantly fewer composite first events (5.32 %/year) than the aspirin group (5.83 %/year; RRR, 8.7 %; p = .043) (Fig. 17.7). The outcome event most dramatically reduced by clopidogrel therapy was MI. There were no major differences between the aspirin and clopidogrel groups in terms of safety. The incidence of significant neutropenia was 0.10 % in the clopidogrel group and 0.17 % in the aspirin group. When patients with coronary disease and either concomitant cerebrovascular disease or peripheral vascular disease were examined, clopidogrel was clearly superior to aspirin in reducing outcome events in this subgroup (RRR, 22.7 %).
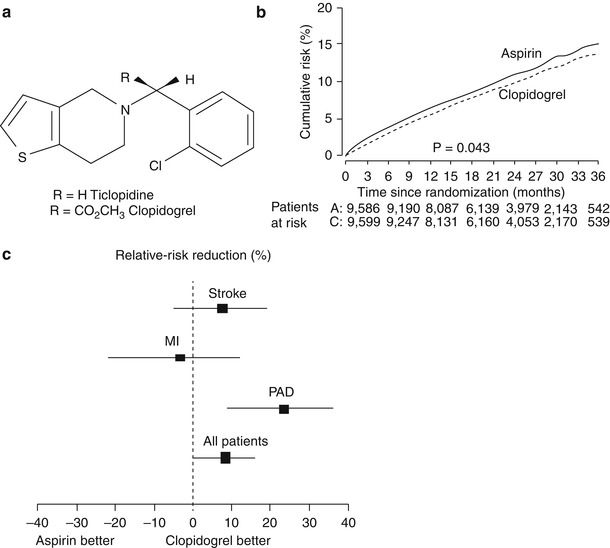
Fig. 17.7
(a) The chemical structure for ticlopidine and its analogue clopidogrel. (b) The slight benefit of clopidogrel over aspirin in the Clopidogrel Versus Aspirin in Patients at Risk of Ischaemic Events (CAPRIE) trial in reducing future myocardial infarction, stroke, and vascular death. (c) The influence of aspirin or clopidogrel in the CAPRIE trial by future event. PAD peripheral artery disease (b, c from Ref. [79]. Reprinted with permission from Elsevier Limited)
Mechanistically, an important factor may be the key role that ADP plays in shear-induced platelet aggregation. In peripheral vascular disease and CAD plus disease in other vascular beds, there is a greater atherosclerotic burden, more shear forces, and, probably, a more important role for ADP-induced platelet activation/aggregation. Additional analyses of the CAPRIE cohort have documented the significant benefit of clopidogrel over aspirin in patients with a history of coronary artery bypass grafts (CABG) and patients with diabetes. These analyses have also shown the benefits of clopidogrel over aspirin in preventing not only initial events (the primary CAPRIE analysis) but also recurrent and total vascular events [80–82].
The Clopidogrel in Unstable Angina to Prevent Recurrent Ischemic Events (CURE) trial was a multicenter, randomized, double-blind, placebo-controlled study that tested combination therapy with aspirin and clopidogrel versus aspirin alone in patients with ACS [83]. A total of 12,562 patients with unstable angina or non-Q-wave MI (within 24 h of their last episode of pain) received 75–325 mg aspirin and then were randomly assigned to receive clopidogrel (300 mg load followed by 75 mg daily) or placebo for 3 months to 1 year. The primary end point was a composite of cardiovascular death, MI, or stroke. The main safety end points were major bleeding (disabling or symptomatic intracranial or intraocular bleeding, or a transfusion of more than 2 units) and life-threatening bleeding (hemoglobin decrease of >5 g/dL, hypotension requiring inotropes, bleeding requiring surgery or transfusion of >4 units of blood, or intracranial bleeding).
Seventy-five percent of the patients enrolled in CURE had unstable angina; 25 % had an elevated enzyme or troponin level; 94 % had an abnormal electrocardiogram; and half had ST-segment deviation. Approximately 30 % of the patients underwent revascularization; the mean follow-up was 9 months. Treatment with the combination of clopidogrel and aspirin was associated with a 20 % relative reduction in the primary endpoint of cardiovascular death, MI, or stroke, largely driven by a 23 % relative reduction in the incidence of MI. Differences in the other components of the primary end point (cardiovascular death, stroke, and non-cardiovascular death) failed to reach statistical significance. The curves for the primary end point began to diverge very early, favoring clopidogrel (within the first few hours). At 24 h, a 20 % relative reduction in the composite of death, MI, and stroke was also noted (Fig. 17.8). The benefits of clopidogrel were present across all major subgroups: patients with and without major ST-segment deviation, enzyme or troponin elevation, or prior and subsequent revascularization [84]. The frequency of composite events with long-term therapy was observed in addition to the in-hospital benefit. Although there was a 34 % excess of major bleeding in the clopidogrel arm, there was no significant excess of life-threatening bleeding with combination therapy.
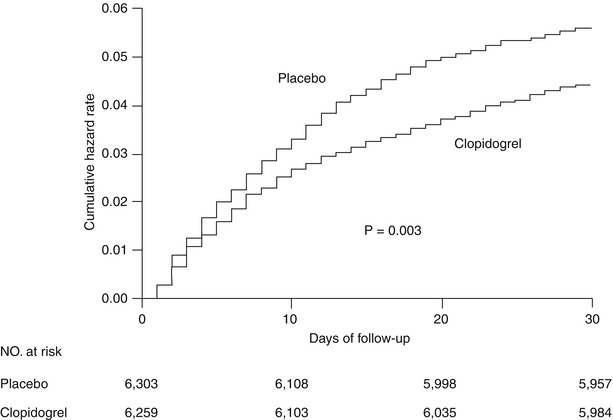
Fig. 17.8
Cumulative hazard rates for the first primary outcome (death from cardiovascular causes, nonfatal myocardial infarction, or stroke) during the first 30 days after randomization of clopidogrel and placebo (plus aspirin) in the CURE trial (From Yusuf et al. [83]. Reprinted with permission from Massachusetts Medical Society)
The promising results of the CURE and CAPRIE trials are a strong argument for the use of clopidogrel in the secondary prevention of atherosclerotic disease. The CURE trial assessed the therapeutic role of clopidogrel, in conjunction with aspirin, immediately after an ischemic event (non-ST-elevation MI), whereas the CAPRIE trial analyzed the benefit of clopidogrel over aspirin in patients with recent MI, stroke, or peripheral arterial disease. These two trials of secondary prevention beg the question of the role of clopidogrel in primary prevention of cardiovascular disease in patients with either a high risk for cardiovascular disease or a remote history of cardiovascular disease. The Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management, and Avoidance (CHARISMA) trial was designed to help address those issues.
In the CHARISMA trial, 15,603 patients with either clinically evident cardiovascular disease or multiple risk factors for cardiovascular disease were randomly assigned to receive either clopidogrel and low-dose aspirin or placebo and low-dose aspirin [85]. The patients were then followed up for a median of 28 months. The primary efficacy end point was a composite of MI, stroke, or death from cardiovascular causes. The frequency of this primary end point was not statistically different between the two groups (6.8 % with clopidogrel plus aspirin vs. 7.3 % with aspirin alone; p = .22) (Table 17.13).
Table 17.13
End points in the CHARISMA trial
End point | Clopidogrel + aspirin (n = 7,802), n (%) | Placebo + aspirin (n = 7,801), n (%) | Relative risk | p value |
|---|---|---|---|---|
Primary end point | 534 (6.8) | 573 (7.3) | 0.93 | 0.22 |
Death from any cause | 371 (4.8) | 374 (4.8) | 0.99 | 0.90 |
Death from any cardiovascular cause | 238 (3.1) | 229 (2.9) | 1.04 | 0.68 |
MI (nonfatal) | 147 (1.9) | 159 (2.0) | 0.92 | 0.48 |
Ischemic stroke (nonfatal) | 132 (1.7) | 160 (2.1) | 0.82 | 0.10 |
Stroke (nonfatal) | 149 (1.9) | 185 (2.4) | 0.80 | 0.05 |
Secondary efficacy end pointa | 1,301 (16.7) | 1,395 (17.9) | 0.92 | 0.04 |
Hospitalization for unstable angina, TIA,or revascularization | 866 (11.1) | 957 (12.3) | 0.90 | 0.02 |
However, a prespecified secondary end point—the composite primary end point plus hospitalizations—did show a moderate significant difference (16.7 % with clopidogrel plus aspirin vs. 17.9 % with aspirin alone; p = .04). There was no significant increase in the rate of severe bleeding (1.7 % with clopidogrel plus aspirin compared to 1.3 % with aspirin alone; p = .09). Moderate bleeding (necessitating blood transfusion) was significantly higher with clopidogrel (2.1 %) than with aspirin alone (1.3 %; p < .01). Looking further at the subgroups of patients included in the study, there appeared to be benefit in patients with “symptomatic atherothrombosis,” and there was a suggestion of harm in patients with only multiple risk factors and no manifest disease (Fig. 17.9). Symptomatic atherothrombosis was defined as documented coronary disease (n = 12,153). In these secondary prevention patients, the primary end point was significantly less frequent with clopidogrel plus aspirin (6.9 %) than with aspirin alone (7.9 %; p = .046). However, in the 3,248 patients with only multiple risk factors and no documented cardiovascular disease (a primary prevention group), there was an increase in the primary end point with clopidogrel plus aspirin (5.4 % vs. 3.8 % with aspirin alone; p = .04) (Table 17.14). The most surprising aspect of this finding is that it held true even in the diabetic patients, who would otherwise be regarded as having a CAD equivalent. Why there should be any potential hazard in this group is unknown.
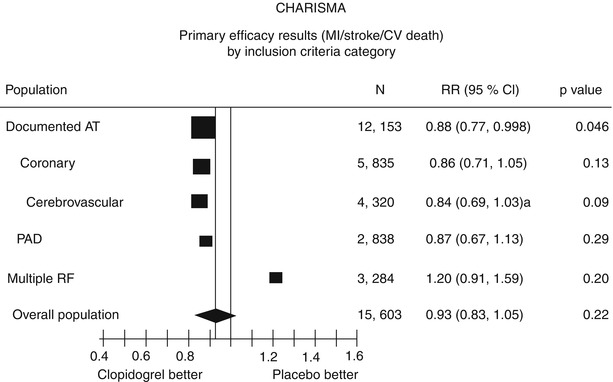
Fig. 17.9
A summary of the results of the CHARISMA trial. The primary efficacy end points are shown by inclusion criteria. Note that in this subgroup analysis, clopidogrel plus aspirin is only significantly superior to aspirin alone in the patients with documented atherothrombotic disease. Furthermore, clopidogrel plus aspirin may be more harmful than aspirin alone in those patients with only risk factors for atherothrombotic disease (From Bhatt et al. [85]. Reprinted with permission from Massachusetts Medical Society)
Table 17.14
Safety end points in the CHARISMA trial
End point | Clopidogrel + aspirin (n = 7,802), n (%) | Placebo + aspirin (n = 7,801), n (%) | Relative risk | p value |
|---|---|---|---|---|
Severe bleeding | 130 (1.7) | 104 (1.3) | 1.25 | 0.09 |
Fatal bleeding | 26 (0.3) | 17 (0.2) | 1.53 | 0.17 |
Primary intracranial hemorrhage | 26 (0.3) | 27 (0.3) | 0.96 | 0.89 |
Moderate bleeding | 164 (2.1) | 101 (1.3) | 1.62 | <0.001 |
The major message from the CHARISMA trial seems to be that clopidogrel plus aspirin is no better than aspirin alone for primary prevention, even in patients with diabetes. For secondary prevention (such as in patients with stable CAD), the message is more ambiguous because this is a secondary subgroup analysis. Clopidogrel plus aspirin appears to provide longer-term benefit than aspirin alone in patients with established disease. This finding, however, will have to be prospectively evaluated if we are to have any hope of risk-stratifying patients for the optimal use of long-term combination therapy.
Two relatively new thienopyridines, prasugrel (Effient) and ticagrelor (Brilinta), may one day replace clopidogrel as the drug of choice for treating stable angina. Prasugrel was approved by both the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA) in 2009 for use in patients who undergo percutaneous coronary intervention (PCI) for unstable angina or MI. Because prasugrel is metabolized more efficiently than clopidogrel, prasugrel inhibits the ADP receptor—and, therefore, platelet activation—more effectively. The PRINCIPLE-TIMI 44 (Prasugrel in Comparison to Clopidogrel for Inhibition of Platelet Activation and Aggregation–Thrombolysis in Myocardial Infarction 44) trial [86] found that in patients undergoing cardiac catheterization for planned PCI, a loading dose of 60 mg prasugrel resulted in much more inhibition of platelet activation (74.8 ± 13.0 %) than a loading dose of 600 mg clopidogrel (31.8 ± 21.1 %) 6 h after administration (p <0.0001). Furthermore, when both drugs were used as maintenance therapy, 10 mg/day prasugrel inhibited platelet activity more effectively than 150 mg/day clopidogrel (61.3 ± 17.8 % vs. 46.1 ± 21.3 %; p <0.0001).
In the TRITON-TIMI 38 trial [87], the effects of prasugrel and clopidogrel were compared in 13,608 patients with ACS who were scheduled to undergo PCI. The results showed that the primary end point (nonfatal MI, nonfatal stroke, or death from a cardiovascular cause) was less frequent in patients who received prasugrel (9.9 %) than in patients who received clopidogrel (12.1 %) [hazard ratio (HR), 0.81; 95 % confidence interval (CI), 0.73–0.90; p <0.001]. In addition, prasugrel was associated with lower rates of MI (7.4 % vs. 9.7 %; p <0.001), urgent target-vessel revascularization (2.5 % vs. 3.7 %; p <0.001), and in-stent thrombosis (1.1 % vs. 2.4 %; p <0.001) than clopidogrel (Table 17.15). However, prasugrel recipients more often had life-threatening bleeding events than clopidogrel recipients (1.4 % vs. 0.9 %, p = 0.01) (Table 17.16).




Table 17.15
TRITON-TIMI 38 trial: major efficacy end points in the overall cohort at 15 monthsa
End point | Prasugrel n = 6,813 (%) | Clopidogrel n = 6,795 (%) | Hazard ratio for prasugrel (95 % CI) | p valueb |
|---|---|---|---|---|
Death from cardiovascular causes, nonfatal MI, or nonfatal strokec | 643 (9.9) | 781 (12.1) | 0.81 (0.73–0.90) | <0.001 |
Death from cardiovascular causes | 133 (2.1) | 150 (2.4) | 0.89 (0.70–1.12) | 0.31 |
Nonfatal MI | 475 (7.3) | 620 (9.5) | 0.76 (0.67–0.85) | <0.001 |
Nonfatal stroke | 61 (1.0) | 60 (1.0) | 1.02 (0.71–1.45) | 0.93 |
Death from any cause | 188 (3.0) | 197 (3.2) | 0.95 (0.78–1.16) | 0.64 |
Death from cardiovascular causes, nonfatal MI, or urgent target-vessel revascularization | 652 (10.0) | 798 (12.3) | 0.81 (0.73–0.89) | <0.001 |
Death from any cause, nonfatal MI, or nonfatal stroke | 692 (10.7) | 822 (12.7) | 0.83 (0.75–0.92) | <0.001 |
Urgent target-vessel revascularization | 156 (2.5) | 233 (3.7) | 0.66 (0.54–0.81) | <0.001 |
Death from cardiovascular causes, nonfatal MI, nonfatal stroke, or rehospitalization for ischemia | 797 (12.3) | 938 (14.6) | 0.84 (0.76–0.92) | <0.001 |
Stent thrombosisd | 68 (1.1) | 142 (2.4) | 0.48 (0.36–0.64) | <0.001 |
From Wiviott et al. [87]. Reprinted with permission from Massachusetts Medical Society
CI confidence interval, MI myocardial infarction
aThe percentages are Kaplan-Meier estimates of the rate of the end point at 15 months. Patients could have had more than one type of end point. Death from cardiovascular causes and fatal bleeding (Table 17.16) are not mutually exclusive, because intracranial hemorrhage and death after cardiovascular procedures that were complicated by fatal bleeding were included in both end points
bp values were calculated with the use of the log-rank test. The prespecified analysis for the primary end point used the Gehan–Wilcoxon test, for which the p value was less than 0.001
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
