Mechanisms of Cardiac Arrhythmias and Conduction Disturbances: Introduction
Recent years have witnessed important advances in our understanding of the molecular and electrophysiologic mechanisms underlying the development of a variety of cardiac arrhythmias (Table 38–1) and conduction disturbances. Progress in our understanding of these phenomena has been fueled by innovative advances in our understanding of the genetic basis and predisposition for electrical dysfunction of the heart. These advances notwithstanding, our appreciation of the basis for many rhythm disturbances is incomplete. This chapter examines our present understanding of cellular, ionic, and molecular mechanisms responsible for cardiac arrhythmias, placing them in historical perspective whenever possible.
| Tachycardia | Mechanism | Origin | AV or VA Conduction |
|---|---|---|---|
| Sinus tachycardia | Automatic (normal) | Sinus node | 1:1 |
| Sinus node reentry | Reentry | Sinus node and right atrium | 1:1 or variable |
| Atrial fibrillation | Reentry, automatic, triggered activity | Atria, thoracic veins, pulmonary veins, SVC, vein of Marshall | Variable |
| Atrial flutter | Reentry | RA, LA (infrequent) | Variable |
| Atrial tachycardia | Reentry, automatic, triggered activity | Atria | 1:1, 2:1, or variable |
| AV nodal reentry tachycardia | Reentry | AV junction | 1:1 or variable |
| AV reentry (WPW or concealed accessory AV connection) | Reentry | Circuit includes accessory AV connection, atria, AV node, His-Purkinje system, ventricles | 1:1 |
| Accelerated AV junctional tachycardia | Automatic | AV junction (AV node and His bundle) | 1:1 or variable |
| Accelerated idioventricular rhythm | Abnormal automaticity | Purkinje fibers | Variable, 1:1, or AV dissociation |
| Ventricular tachycardia | Reentry, automatic, triggered | Ventricles | AV dissociation, variable |
| Bundle branch reentrant tachycardia | Reentry | Bundle branches and ventricular septum | AV dissociation, variable, or 1:1 |
| RVOT | Automatic, triggered activity | RVOT | AV dissociation, variable, or 1:1 |
| TdP tachycardia | Reentry, triggered activity | Ventricles | AV dissociation |
| Bidirectional tachycardia | Triggered activity | Purkinje cells | AV dissociation |
Arrhythmic activity can be categorized as passive (eg, atrioventricular [AV] block) or active. The mechanisms responsible for active cardiac arrhythmias are generally divided into two major categories: (1) enhanced or abnormal impulse formation and (2) reentry (Fig. 38–1). Reentry occurs when a propagating impulse fails to die out after normal activation of the heart and persists to reexcite the heart after expiration of the refractory period. Evidence implicating reentry as a mechanism of cardiac arrhythmias stems back to the turn of century.1-3 Multichannel mapping studies documented that reentrant wavefronts may underlie the mechanisms of atrial4 and ventricular5 tachyarrhythmias. Phase 2 reentry,6 spiral waves of excitation,7 and fibrillatory conduction8 are interesting concepts of reentrant activity advanced to explain the development of extrasystolic activity and atrial as well as ventricular fibrillation (VF). Mechanisms responsible for abnormal impulse formation include enhanced automaticity and triggered activity. Automaticity can be further subdivided into normal and abnormal. Triggered activity consists of (1) early afterdepolarizations (EADs), (2) late-phase 3 EAD, and (3) delayed afterdepolarizations (DADs).9-11 Although traditionally automaticity and triggered activity are thought to be two distinct mechanisms of arrhythmogenesis, more recent studies suggest that they might share the same mechanism.
Figure 38–1.
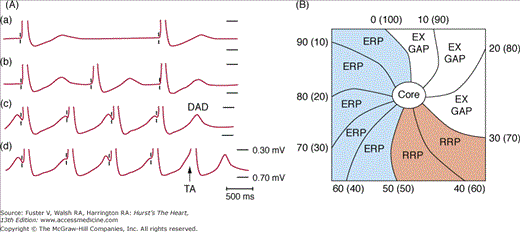
Triggered activity and reentry. A. Pacing-induced delayed afterdepolarization (DAD) in simian mitral valve. Progressively increased pacing rates (from A to D) induced an increased size of DADs and triggered activity (TA) probably caused by intracellular Ca accumulation. B. A schematic of reentrant excitation. Reentry can be either functional or anatomical. Both types of reentry can be associated with an excitable gap (EX GAP), a relative refractory period (RRP), and an effective refractory period (ERP). The numbers represent the times (ms) of activation, and the numbers in the parenthesis are the times (ms) of recovery. Part A From Wit et al9 with permission. Part B reproduced with permission from Bonometti et al.253
Abnormal Impulse Formation
Automaticity is the property of cardiac cells to generate spontaneous action potentials. Spontaneous activity is the result of diastolic depolarization caused by a net inward current during phase 4 of the action potential, which progressively brings the membrane potential to threshold. The sinoatrial (SA) node normally displays the highest intrinsic rate. All other pacemakers are referred to as subsidiary or latent pacemakers because they take over the function of initiating excitation of the heart only when the SA node is unable to generate impulses or when these impulses fail to propagate. There is a hierarchy of intrinsic rates of subsidiary pacemakers that have normal automaticity: atrial pacemakers have faster intrinsic rates than AV junctional pacemakers, and AV junctional pacemakers have faster rates than ventricular pacemakers.
Wit and Cranefield10 defined automatic activity as activity that arises in the absence of an external cause (ie, activity that does not have to be triggered by a stimulated action potential). The prototypical example of automaticity is the spontaneous beating of the SA node. On the other hand, triggered activity was the activity in which nondriven action potentials were initiated by one or more driven action potentials. The authors used the term triggered to explain the mechanisms by which quiescent fibers remained quiescent until driven at a fast rate. However, recent studies have shown that automaticity and triggered activity may share a common mechanism that is, activation of the sodium-calcium exchange current (INCX).12
Lakatta, Maltsev, and their collaborators13-16 used the terms sarcolemma voltage clocks and the subsarcolemmal Ca clocks to describe the mechanisms of SA node automaticity. The voltage clock is formed by voltage-sensitive membrane currents, such as the hyperpolarization-activated pacemaker current (If).17,18 This current is also referred to as a “funny“ current because, unlike the majority of voltage-sensitive currents, it is activated by hyperpolarization (from -40/-50 mV to -100/-110 mV) rather than depolarization. At the end of the action potential, the If is activated and depolarizes the sarcolemmal membrane.19,20 The If is a mixed Na-K inward current modulated by the autonomic nervous system through cAMP.21 Sympathetic stimulation (isoproterenol) increases the If, and parasympathetic stimulation (acetylcholine) reduces If.22 These findings suggest that If is responsible for heart rate control by the autonomic nervous system. The depolarization activates ICa,L, which provides Ca to activate the cardiac ryanodine receptor (RyR2). The activation of RyR2 initiate sarcoplasmic reticulum (SR) Ca release (Ca-induced Ca release), leading to contraction of the heart, a process known as EC coupling. The intracellular Ca (Cai) is then pumped back into SR by the SR Ca-ATPase (SERCA2a) and completes this Ca cycle. In addition to If, multiple time- and voltage-dependent ionic currents have been identified in cardiac pacemaker cells, which contribute to diastolic depolarization. These currents include (but are not limited to) ICa-L, ICa-T, IST, and various types of delayed rectifier K currents.23 Many of these membrane currents are known to respond to β-adrenergic stimulation. All of these membrane ionic currents contribute to the regulation of SA node automaticity by changing the membrane potential.
The If is not the only depolarizing current active in late phase 3 or phase 4 of the action potential. Another important ionic current that can depolarize the cell is the sodium-calcium exchanger current (INCX). In its forward mode, the INCX exchanges three extracellular Na+ with one intracellular Ca2+, resulting in a net intracellular charge gain. This electrogenic current is active during late phase 3 and phase 4 because the Cai decline outlasts the SA node action potential duration (APD). Recent studies have shown that INCX may participate in normal pacemaker activity.24–29 The sequence of events includes spontaneous rhythmic SR Ca release, Cai elevation, the activation of INCX, and membrane depolarization.30 This process is highly regulated by the cAMP and the autonomic nervous system.31 According to their findings, sympathetic stimulation accelerates heart rate by phosphorylation of proteins that regulate Cai balance and spontaneous SR Ca cycling. These proteins include phospholamban (PLB, an SR membrane protein regulator of SERCA2a), L-type Ca channels, and RyR2. Phosphorylation of these proteins controls the phase and size of subsarcolemmal SR Ca releases. The resultant INCX is crucial for both basal and reserve cardiac pacemaker function.
Many of the elegant studies on automaticity were performed in isolated SA node cells. However, the SA node is a complex structure32-36 and that many factors interact with each other to ensure the initiation of the heart beats. Activation maps in intact canine right atria (RA) have shown that the SA node impulse origin is multicentric,37 and sympathetic stimulation predictably results in a cranial (superior) shift of the pacemaking site in humans and dogs.38,39 Based on evidence from isolated SA node myocytes, late diastolic Cai elevation before the membrane action potential upstroke is a key signature of pacemaking by the Ca clock. Simultaneous mapping of membrane potential and Cai transient in Langendorff-perfused canine RA preparation40 showed changes consistent with Ca clock mechanism of impulse generation (Fig. 38–2). In that study, sympathetic stimulation (isoproterenol) induced spontaneous SR Ca release during phase 4 depolarization of intact canine SA node. The spontaneous Ca release then induces upward (cranial) shift of the leading pacemaker site and heart rate acceleration. Because the leading pacemaker site is not in a fixed location, radiofrequency catheter ablation of the SA node can be a difficult procedure.
Figure 38–2.
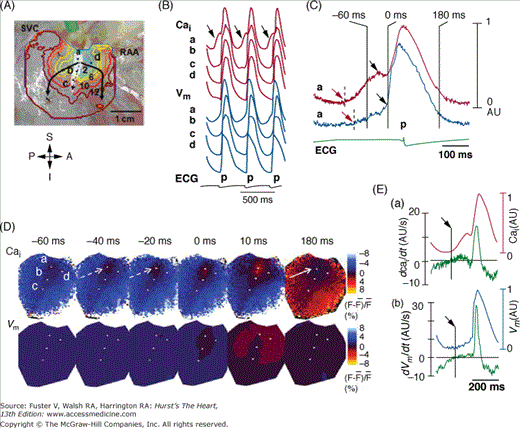
Activation pattern of sinoatrial (SA) node and surrounding right atrium (RA) during isoproterenol infusion of 0.3 μmol/L. This study was done in isolated Langendorff-perfused RA preparation. A. Isochronal map of Vm. The number on the each isochronal line indicates time (ms). The white shaded area is the SA node identified by the presence of spontaneous phase 4 depolarization. B. The Vm (blue) and Cai (red) recordings from the superior (a), middle (b), and inferior (c) SA node and RA (d) presented in A. C. Magnified view of Cai and Vm tracings of superior SA node. Note the robust late diastolic Ca elevation before phase 0 of action potential (0 ms), which in turn was much earlier than onset of the p wave on electrocardiography. D. The Vm and Cai ratio maps at times from –60 ms before to 180 ms after phase 0 AP of C. The late diastolic Ca2+ elevation (broken arrows in frame –40 and –20 ms) was followed by the Cai sinkhole during early diastole (solid arrow in frame 180 ms). E. (a) Cai and dCai/dt. (b) Vm and dVm/dt. The onset of late diastolic Ca2+ elevation and diastolic depolarization were (arrows) identified by the time when dCai/dt and dVm/dt, respectively, crossed the baseline. RAA, right atrial appendage; SVC, superior vena cava. Reproduced with permission from Joung et al.40
In addition to the SA node, AV nodes and the Purkinje system are also capable of generating automatic activity. The contribution of If and IK differs in SA node or AV nodes and Purkinje fiber because of the different potential ranges of these two pacemaker types (ie, –70 to –35 mV and –90 to –65 mV, respectively). The contribution of other voltage-dependent currents can also differ among the different cardiac cell types. Whether or not Ca clock plays a role in pacemaking of AV node and Purkinje cells remain unclear. Cells in the SA node possess the fastest intrinsic rates. Thus, the SA node is the primary pacemaker in the normal heart. When impulse generation or conduction within or out of the SA node is impaired, latent or subsidiary pacemakers within the atria or ventricle are capable of taking control of pacing the heart. The intrinsically slower rates of these latent pacemakers result in bradycardia. Subsidiary atrial pacemakers with more negative diastolic potentials (–75 to –70 mV) than SA nodal cells are located at the junction of the inferior RA and the inferior vena cava, near or on the Eustachian ridge.41,42 Other atrial pacemakers have been identified in the crista terminalis43 as well as at the orifice of the coronary sinus9 and in the atrial muscle that extends into the tricuspid and mitral valves.42,43 The cardiac muscle sleeves that extend into the cardiac veins (venae cavae and pulmonary veins) can also have the property of normal automaticity.44 Latent pacemaking cells in the AV junction are responsible for AV junctional rhythms.45 Both atrial and AV junctional subsidiary pacemakers are under autonomic control, with the sympathetic system increasing and parasympathetic system slowing the pacing rate.
The slowest subsidiary pacemakers are found in the His-Purkinje system in the ventricles of the heart. In the His-Purkinje system, parasympathetic effects are less apparent than those of the sympathetic system. Although acetylcholine produces little in the way of a direct effect, it can significantly reduce Purkinje automaticity by means of the inhibition of the sympathetic influence, a phenomenon termed accentuated antagonism.46-48 Simultaneous recording of cardiac sympathetic and parasympathetic activity in ambulatory dogs confirmed that sympathetic activation followed by vagal activation may be associated with significant bradycardia.49,50 Accentuated antagonism is also important in abnormal automaticity in the His-Purkinje system.51 Adenosine has no effects on His-Purkinje system activation rate at baseline but reduces its activation rate during isoproterenol infusion. These findings suggest that adenosine's effects on the human His-Purkinje system are primarily antiadrenergic and are thus consistent with the concept of accentuated antagonism. These effects of adenosine may serve as a counterregulatory metabolic response that improves the O2 supply–demand ratio perturbed by enhanced sympathetic tone. Some catecholamine-mediated ventricular arrhythmias that occur during ischemia or enhanced adrenergic stress may be caused by an imbalance in this negative feedback system.
Changes of ionic component of cardiac cells by gene transfer can convert ordinary myocytes into cardiac pacemakers. One approach is to insert hyperpolarization-activated cyclic nucleotide-gated (HCN) channel into cells using viral gene transfer, cell fusion, or stem cell technologies.52-54 Successful HCN expression in the myocytes leads to the activation of pacemaker current (If), enabling nonpacemaking cells to generate automaticity. These studies further confirm the importance of If in the mechanisms of automaticity.
Abnormal automaticity includes both reduced automaticity, which causes bradycardia, and increased automaticity, which causes tachycardia. Arrhythmias caused by abnormal automaticity can result from diverse mechanisms (see Table 38–1). Alterations in sinus rate can be accompanied by shifts of the origin of the dominant pacemaker within the sinus node or to subsidiary pacemaker sites elsewhere in the atria. Impulse conduction out of the SA mode can be impaired or blocked as a result of disease or increased vagal activity, leading to development of bradycardia. AV junctional rhythms occur when AV junctional pacemakers located either in the AV node or in the His bundle accelerate to exceed the rate of SA node or when the SA nodal activation rate was too slow to suppress the AV junctional pacemaker.
Bradycardia can occur in structurally normal hearts because of genetic mutations that result in abnormalities of either membrane clock or Ca clock mechanism of automaticity. One example is the mutation of HCN4, which is part of the channels that carry If. Mutations of the HCN4 cause familial bradycardia.55-57 However, the bradycardia caused by HCN4 mutations may be entirely asymptomatic. Although HCN4 mutations cause baseline bradycardia, the heart rate responses to exercise may be either suboptimal with a maximum rate of 100 beats/min56 or entirely normal with maximum rates of more than 150 beats/min.57 The presence of normal heart rate response cannot be explained by the HCN4 mutation in those patients. Therefore, although these findings support the importance of If in generating SA node automaticity in humans, the exercise-induced heart rate acceleration may be attributable to a different mechanism. The most likely mechanism is increased activity of the Ca clocks by phosphorylation of PLB and by increased activity of ICa,L and RyR2 during sympathetic stimulation. Normal SR Ca release depends on a complex formed by calsequestrin (CSQ), RyR2, junctin, and triadin.58 Mutations of RyR2 and CSQ increase SR Ca release and cause catecholaminergic polymorphic ventricular tachycardia (CPVT).59-61 Although tachycardia is the dominant symptomatic phenotype, it is interesting to note that patients with CPVT also exhibit significant bradycardia.62 The association of bradycardia with Ca-handling abnormalities is consistent with the hypothesis that Ca clock is important in the mechanisms of SA node automaticity. However, further investigation is needed to establish a causal relationship between Ca clock and bradycardia in patients with CPVT.
Common diseases, such as heart failure and atrial fibrillation (AF), may be associated with significant SA node dysfunction.63,64 Malfunction of both membrane voltage clocks and Ca clocks might be present in both of these common diseases. Zicha et al65 reported downregulation of HCN4 expression contributes to heart failure–induced sinus node dysfunction,64 and upregulation of atrial HCN4 may help to promote atrial arrhythmia formation. Heart failure is also known to be associated with significant abnormalities of Cai regulation.66,67 It is likely that abnormalities of both membrane voltage clock and Ca clock are responsible for the SA node dysfunction in heart failure.64 AF is also associated with a downregulation of If in canine models.68 However, AF can also be associated with Ca clock malfunction.40 These studies show that as in heart failure, SA node dysfunction in AF is also associated with malfunction of both membrane and Ca clocks. The SA node malfunction is reversible after successful radiofrequency catheter ablation of AF.69
Atrial and ventricular myocardial cells do not display spontaneous diastolic depolarization or automaticity under normal conditions, but they can develop these characteristics when depolarized, resulting in the development of repetitive impulse initiation, a phenomenon termed depolarization-induced automaticity.70 The membrane potential at which abnormal automaticity develops ranges between –70 and –30 mV. The rate of abnormal automaticity is substantially higher than that of normal automaticity and is a sensitive function of resting membrane potential (ie, the more depolarized resting potential, the faster the rate). Similar to normal automaticity, abnormal automaticity is enhanced by β-adrenergic agonists and by reduction of external potassium. Depolarization of membrane potential associated with disease states is most commonly a result of (1) an increase in extracellular potassium, which reduces the reversal potential for IK1, the outward current that largely determines the resting membrane or maximum diastolic potential; (2) a reduced number of IK1 channels; (3) a reduced ability of the IK1 channel to conduct potassium ions; or (4) electrotonic influence of neighboring depolarized zone. Because the conductance of IK1 channels is a sensitive to extracellular potassium concentration, hypokalemia can lead to major reduction in IK1, leading to depolarization and the development of enhanced or abnormal automaticity, particularly in Purkinje pacemakers. A reduction in IK1 can also occur secondary to a mutation in KCNJ2, the gene that encodes for this channel, leading to increased automaticity and extrasystolic activity presumably arising from the Purkinje system.71,72 Interestingly, because β-adrenergic stimulation is effective in augmenting IK1,73 sympathetic stimulation can produce a paradoxical slowing of automaticity and ectopy in this setting.
Normal or subsidiary pacemaker activity can also be enhanced, leading to sinus tachycardia or a shift to ectopic sites within the atria, giving rise to atrial tachycardia. One cause can be enhanced autonomic nerve activity. Direct recording from the stellate ganglion and vagal nerves show that stellate ganglion nerve activity (SGNA) and vagal nerve activity (VNA) are both important in controlling sinus rate and in triggering atrial tachycardia in ambulatory dogs. Figure 38–3 shows an example of heart rate acceleration induced by SGNA. The SGNA consists of two types of activity. The vast majority of activity was in the form of low-amplitude burst discharge activity (LABDA). A second form of activity is the high-amplitude spike discharge activity (HASDA), which occurs less than 10 times daily in normal dogs; the incidence may more than double in dogs with heart failure or myocardial infarction (MI).49,50,74 In this and in all dogs studied, there was no latency between the onset of nerve activity and the shortening of the next PP interval. Vagal nerve activation, on the other hand, induces sinus bradycardia.49Figure 38–4 shows an example of bradycardia associated with increased vagal nerve activity. The same figure shows that simultaneous sympathovagal discharges are common triggers of atrial tachyarrhythmia.49
Figure 38–3.
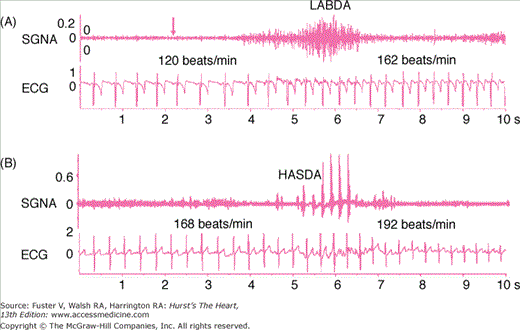
Sympathetic control of heart rate. Stellate ganglion nerve activity (SGNA) was recorded simultaneously with electrocardiogram (ECG) in a normal ambulatory dog with an implanted radiotransmitter. A. Low-amplitude burst discharge activity (LABDA), which induced heart rate acceleration. B. High-amplitude spike discharge activity (HASDA) that occurred during LABDA. HASDA further accelerated the heart rate. The unit for SGNA and ECG is mV. Reproduced with permission from Zhou et al.74
Figure 38–4.
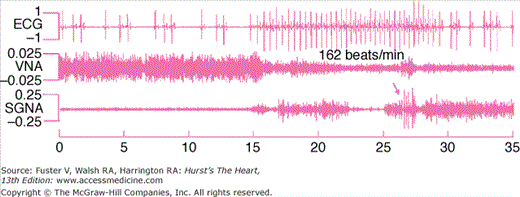
Vagal control of heart rate. This figure was obtained from a normal ambulatory dog with implanted transmitter. Vagal nerve activity (VNA) is associated with slow heart rate response (left part of the figure), and sympathovagal co-activation was associated with atrial tachycardia followed by irregular heart beats and sinus bradycardia toward the end of the tracing. Arrows point to an episode of high-amplitude spike discharge activity (HASDA) amid continuous low-amplitude burst discharge activity (LABDA). Reproduced with permission from Ogawa et al.49
The automaticity of all pacemakers within the heart is inhibited when they are overdrive paced.75 This inhibition is called overdrive suppression. Under normal conditions, all subsidiary pacemakers are overdrive suppressed by SA nodal activity. A possible mechanism of overdrive suppression is intracellular accumulation of Na leading to enhanced activity of the sodium pump (sodium-potassium adenosine triphosphatase [Na+-K+ ATPase]), which generates a hyperpolarizing electrogenic current that opposes phase 4 depolarization.76 The faster the overdrive rate or the longer the duration of overdrive, the greater the enhancement of sodium pump activity, so that the period of quiescence after cessation of overdrive is directly related to the rate and duration of overdrive.
Latent pacemakers throughout the heart are generally reset by the propagating wavefront initiated by the dominant pacemaker and are therefore unable to activate the heart. An exception to this rule occurs when the pacemaking tissue is protected from the impulse of sinus origin. A region of entrance block arises when cells exhibiting automaticity are surrounded by ischemic, infarcted, or otherwise compromised cardiac tissues that prevent the propagating wave from invading the focus but that permit the spontaneous beat generated within the automatic focus to exit and activate the rest of the myocardium. A pacemaker region exhibiting entrance block and exit conduction defines a parasystolic focus. The ectopic activity generated by a parasystolic focus is characterized by premature ventricular complexes with variable coupling intervals, fusion beats, and interectopic intervals that are multiples of a common denominator.
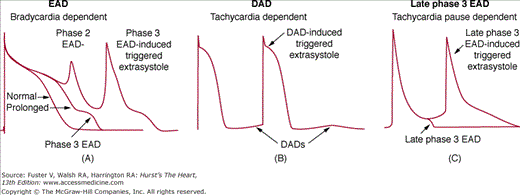
Oscillatory depolarizations that attend or follow the cardiac action potential and depend on preceding transmembrane activity for their manifestation are referred to as afterdepolarizations. Two subclasses traditionally recognized early and delayed. Whereas EADs interrupt or retard repolarization during phase 2 or phase 3 (or both) of the cardiac action potential, DADs occur after full repolarization. When EAD or DAD amplitude suffices to bring the membrane to its threshold potential, a spontaneous action potential referred to as a triggeredresponse is the result. These triggered events can be responsible for extrasystoles and tachyarrhythmias that develop under conditions predisposing to the development of afterdepolarizations.
EADs are observed in isolated cardiac tissues exposed to injury, altered electrolytes, hypoxia, acidosis, catecholamines, and pharmacologic agents (including antiarrhythmic drugs). Ventricular hypertrophy and heart failure also predispose to the development of EADs.77 EAD characteristics vary as a function of animal species, tissue or cell type, and the method by which the EAD is elicited. Although specific mechanisms of EAD induction can differ, a critical prolongation of repolarization accompanies most, but not all, EADs. Drugs that block potassium currents may reduce repolarization reserve and predispose the cells to EADs.78 Figure 38–5 illustrates the two types of EAD generally encountered in Purkinje fibers. Oscillatory events appearing at potentials positive to –30 mV are generally referred to as phase 2 EADs. Those occurring at more negative potentials are termed phase 3 EADs. Phase 2 and phase 3 EADs sometimes appear in the same preparation.
EAD-induced triggered activity is sensitive to stimulation rate. Antiarrhythmic drugs with class III action generally induce EAD activity at slow stimulation rates and totally suppress EADs at rapid rates.79,80 In contrast, β-adrenergic agonist–induced EADs are fast rate dependent.81,82 In the presence of rapid outward potassium current (IKr) blockade, β-adrenergic agonists, or acceleration from an initially slow rate transiently facilitates the induction of EAD activity in ventricular M cells but not in epicardium or endocardium and rarely in Purkinje fibers.83 This biphasic effect is thought to be caused by an initial priming of INCX, which provides an electrogenic inward current that facilitates EAD development and prolongs APD. This early phase is followed by recruitment of slowly activating delayed rectifier current (slow outward potassium current [IKs]), which abbreviates APD and suppresses EAD activity.
EADs develop more in midmyocardial M cells and Purkinje fibers than in epicardial or endocardial cells when exposed to APD-prolonging agents. This is because of the presence of a weaker IKs in M cells.84 Block of IKs with chromanol 293B permits the induction of EADs in canine epicardial and endocardial tissues in response to IKr blockers such as E-4031 or sotalol.85 The predisposition of cardiac cells to the development of EADs depends principally on the reduced availability of IKr and IKs as occurs in many forms of cardiomyopathy. Under these conditions, EADs can appear in any part of the ventricular myocardium.
An EAD occurs when the balance of current active during phase 2 or 3 of the action potential shifts in the inward direction. If the change in current–voltage relation results in a region of net inward current during the plateau range of membrane potentials, it leads to a depolarization or EAD. Most pharmacologic interventions or pathophysiologic conditions associated with EADs can be categorized as acting predominantly through one of four different mechanisms: (1) a reduction of repolarizing potassium currents (IKr, class IA and III antiarrhythmic agents; IKs, chromanol 293B or IK1); (2) an increase in the availability of calcium current (Bay K 8644, catecholamines); (3) an increase in the sodium-calcium exchange current (INCX) caused by augmentation of Cai activity or upregulation of the INCX; or (4) an increase in late sodium current (late INa) (aconitine, anthopleurin-A, and ATX-II). Combinations of these interventions (ie, calcium loading and IKr reduction) or pathophysiologic states can act synergistically to facilitate the development of EADs.
A sustained component of sodium channel current (INa) active during the action potential plateau, originating from channels that fail to inactivate and a non-equilibrium component arising from channels recovering from inactivation during phases 2 and 3, has been shown to contribute prominently the APD and induction of EADs.86,87 Reactivation of ICa,L may contribute to the development of EADs when there is persistent late INa and reduced repolarization reserve.86 Calcium–calmodulin kinase II (CaMKII) has been linked to the development of EAD and torsade de pointes in cellular models in which repolarization is prolonged.88 Ranolazine, which suppresses late INa, is a promising antiarrhythmic agent, especially in the atria.89
DADs occur because of the spontaneous SR Ca release, which activates the INCX and results in oscillations of transmembrane potentials that occur after full repolarization of the action potential.90,91 When there is spontaneous SR Ca release, it activates the INCX and depolarizes the cell membrane. When DADs reach the threshold potential, they give rise to spontaneous action potentials generally referred to as triggered activity. However, whether or not spontaneous SR Ca releases can cause a DAD depends in part on the cell type. Maruyama et al92 used the term diastolic Cai-voltage coupling gain to describe membrane potential (Vm) responses to elevated Cai during diastole. This concept is based on the finding that the same magnitude of SR Ca release may induce DADs in the Purkinje fibers but not in the epicardial cells. The reduced IK1 in the Purkinje cells93 was thought to underlie the higher diastolic Cai-voltage coupling gain than other myocardial cells. As discussed above, recent studies suggest that rhythmic spontaneous SR Ca release (Ca clock) is also a mechanism of SA nodal automaticity. Therefore, although spontaneous SR Ca release is an arrhythmogenic mechanism, it also contributes significantly to the normal automaticity.
Role of Delayed Afterdepolarization-Induced Triggered Activity in the Development of Cardiac Arrhythmias
An excellent example of DAD-induced arrhythmia is the catecholaminergic polymorphic ventricular tachycardia (CPVT), which may be caused by the mutation of either the type 2 ryanodine receptor (RyR2) or the calsequestrin (CSQ2).59,61 The fundamental mechanism of these arrhythmias is the “leaky“ ryanodine receptor, which is aggravated during catecholamine stimulation. A typical clinical phenotype of CPVT is bidirectional ventricular tachycardia (VT), which is also seen in digitalis toxicity.94,95 Because the genetic defects of CPVT has been well described, it is possible to generate transgenic mice that duplicates the human genotype. One of the transgenic mice (RyR2/RyR2(R4496C)) was subjected to optical mapping studies. The isolated cells were studied for DADs. The results showed that these mice exhibited both polymorphic VT and bidirectional VT and hence reproduced the most important phenotypes of CPVT. The in vitro studies documented that DADs underlie both polymorphic and bidirectional VT of these mice. In another study, mice carrying a human D307H missense mutation of calsequestrin (CASQ307/307)96 also demonstrated frequent and prolonged SR Ca release events. If DADs and triggered activity underlie arrhythmias in CPVT syndrome, it follows that inhibition of spontaneous SR Ca release from the RyR2 might lead to successful mechanism-based therapy of cardiac arrhythmia. Wehrens et al97 demonstrated that heterozygous mutation of FKBP12.6 leads to leaky RyR2 and exercise-induced VT and VF, simulating the human CPVT phenotype. RyR2 stabilization with a derivative of 1,4-benzothiazepine (JTV519) increased the affinity of calstabin2 for RyR2, which stabilized the closed state of RyR2 and prevented the Ca leak that triggers arrhythmias. The authors postulated that enhancing the binding of calstabin2 to RyR2 may be a therapeutic strategy for common ventricular arrhythmias. However, JTV519 is not approved for human use. Knollmann et al98 subsequently discovered that flecainide, an antiarrhythmic drug approved by the Food and Drug Administration, prevented adrenergic stress–induced arrhythmias in a mouse model of CPVT and in humans with CASQ2 or RYR2 mutations. The latter study provided first human data that support the efficacy of RyR2-based therapy in human patients. Taken together, these human and transgenic model studies suggest that spontaneous SR Ca release, DAD, and triggered arrhythmias underlie the mechanisms of CPVT. Other studies indicate that DAD-induced extrasystoles serve to trigger catecholamine-induced VT or VF but that the epicardial origin of these ectopic beats increases transmural dispersion of repolarization, thus providing the substrate for the development of reentrant tachyarrhythmias, which underlie the rapid polymorphic VT or VF.99 In addition to CPVT, DADs may also contribute significantly to arrhythmogenesis in common arrhythmias, such as premature ventricular contractions from the peri-infarct zone in rabbit hearts with subacute MI.100 Heart failure is associated with structural and electrophysiologic remodeling, leading to tissue heterogeneity that enhances arrhythmogenesis and the propensity of sudden cardiac death.101–103 The mechanisms of arrhythmogenesis in heart failure may be attributed to the upregulation of INCX activity, abnormal Cai handling, and nonreentrant ventricular arrhythmias caused by triggered activity.101 DADs occur in heart failure because of SR Ca overload and spontaneous SR Ca release that activates electrogenic INCX during phase 4 of the action potential.104 Because DADs are often induced by rapid activation and Cai accumulation, the best time to observe DADs is at the cessation of rapid pacing or tachycardia such as immediately after ventricular defibrillation.92 Ogawa et al105 performed simultaneous optical mapping of Cai and Vm in rabbit hearts with chronic pacing-induced heart failure. Immediately after successful defibrillation, there was spontaneous SR Ca release manifested by Cai elevation before the onset of the action potential. These ventricular premature contractions may initiate recurrent spontaneous VF (VF storm). Figure 38–6 shows an example of spontaneous VF in a Langendorff perfused failing heart. The heart failure was induced by rapid ventricular pacing at 350 beats/min for 3 weeks.105 Part A shows baseline recording. The arrow in B shows spontaneous Cai elevation associated with a DAD on the epicardium.
Figure 38–6.
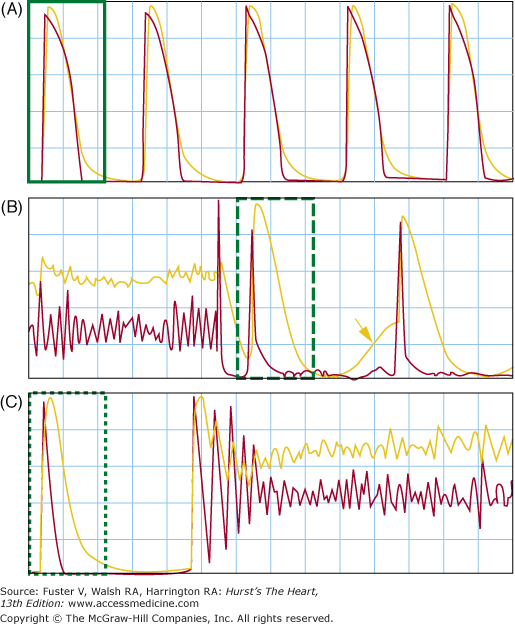
Delayed afterdepolarization (DAD) and late phase 3 early afterdepolarization (EAD) in the postshock period. The figure shows optical mapping of membrane potential (Vm, red line) and intracellular Ca2+ (Cai, yellow line). A. The baseline recording, where the action potential duration and Cai transient were of similar duration. B. Spontaneous Cai elevation after successful defibrillation (arrow) associated with an epicardial DADs on the Vm tracing. That DAD did not induce triggered activity from the epicardium. Subsequent beat originated from outside of the mapped region and is associated with very short action potential duration. C. An episode of spontaneous ventricular fibrillation in the postshock period. Because of the short action potential duration (APD) and long Cai transient, the mechanism of DAD is most consistent with the late phase 3 EAD. Reproduced with permission from Ogawa et al.105
Parts B and C in Fig. 38–6 show Vm and Cai at termination and at the onset of spontaneous VF, respectively. Note the presence of short APD in the immediate postshock period (B) and that the first ectopic beat that initiated VF occurred from late phase 3 of the preceding action potential (C). The mechanism of VF is best explained by the late phase 3 EAD and triggered activity.11,105,106 This arrhythmogenic mechanism combines properties of both EAD and DAD but has its own unique character. Late phase 3 EAD-induced triggered extrasystoles represent a new concept of arrhythmogenesis in which abbreviated repolarization permits normal SR calcium release to induce an EAD-mediated closely coupled triggered response, particularly under conditions permitting intracellular calcium loading. These EADs are distinguished by the fact that they interrupt the final phase of repolarization of the action potential (late phase 3). In contrast to previously described DAD or intracellular calcium (Cai)-dependent EAD, it is normal, not spontaneous, SR Ca release that is responsible for the generation of the EAD. Late phase 3 EADs are observed when APD is markedly abbreviated as with acetylcholine (Figs. 38–5C and 38–6B). Based on the time course of contraction, levels of Cai would be expected to peak during the plateau of the action potential (membrane potential,~−5 mV) under control conditions but during the late phase of repolarization (membrane potential, ~−70 mV) in the presence of acetylcholine. As a consequence, the two principal Ca-mediated currents, INCX and ICl(Ca), would be expected to be weakly inward or even outward (ICl(Ca)) when APD is normal (control) but strongly inward when APD is very short (such as during acetylcholine infusion).11 Thus, abbreviation of the atrial APD allows for a much stronger recruitment of both INCX and ICl(Ca) in the generation of a late phase 3 EADs. In the isolated canine atria, late phase 3 EAD-induced extrasystoles have been shown to initiate AF, particularly after spontaneous termination of the arrhythmia (IRAF, immediate reinduction of AF).11 Patterson and colleagues107 described “tachycardia-pause“-induced EAD in isolated superfused canine pulmonary vein muscular sleeve preparations in the presence of both simultaneous parasympathetic (to abbreviate APD) and sympathetic (to augment Cai) nerve stimulation. This EAD also appears during late phase 3 of the action potential, and a similar mechanism has been proposed.107 Simultaneous sympathovagal activation is also known to be the primary trigger of paroxysmal atrial tachycardia and AF episodes in dogs with intermittent rapid pacing.50 In addition to the atrial arrhythmias, late phase 3 EAD may also be responsible for the development recurrent VF in failing hearts.105 As shown in Fig. 38–6, Langendorff perfused failing hearts develop acute and transient APD shortening immediately after successful defibrillation. Because of Cai accumulation during fibrillation, a combination of short APD and large Cai allows late phase 3 EAD to occur. Therefore, in addition to recurrent atrial arrhythmias, late phase 3 EADs may also be the mechanism responsible for ventricular arrhythmias associated with heart failure.
Reentrant Arrhythmias
Reentry is a fundamentally different mechanism of arrhythmogenesis than automaticity or triggered activity. The circus movement reentry occurs when an activation wavefront propagates around an anatomical or functional core and reexcites the site of origin. In this type of reentry, all cells take turns to recover from excitation and ready to be excited again when the next wavefront arrives. In comparison, reflection and phase 2 reentry occur when there are large differences of recovery from one site to another. The site with delayed recovery serves as a virtual electrode that excites its already recovered neighbor, resulting in reentry. A circus movement is not observed. In addition, reentry can also be classified as anatomical and functional, although there is a grey zone in which both functional and anatomical factors are important in determining the characteristics of reentrant excitation.
The ring model is the prototypical example of reentry around an anatomical obstacle. It first emerged as a concept shortly after the turn of the last century when Mayer reported the results of experiments involving the sub-umbrella tissue of a jellyfish (Sychomedusa cassiopeia).1 The muscular disk did not contract until ringlike cuts were made and pressure and a stimulus applied. This caused the disc to “spring into rapid rhythmical pulsation so regular and sustained as to recall the movement of clockwork.“ Mayer demonstrated similar circus movement excitation in rings cut from the ventricles of turtle hearts, but he did not consider this to be a plausible mechanism for the development of cardiac arrhythmias. His experiments proved valuable in identifying two fundamental conditions necessary for the initiation and maintenance of circus movement excitation: (1) unidirectional block (the impulse initiating the circulating wave must travel in one direction only) and (2) for the circus movement to continue, the circuit must be long enough to allow each site in the circuit to recover before the return of the circulating wave. GR Mines3 was the first to develop the concept of circus movement reentry as a mechanism responsible for cardiac arrhythmias. He confirmed Mayer' observations and suggested that the recirculating wave could be responsible for clinical cases of tachycardia.108 The clinical importance of the reentry was reinforced with the discovery by Kent of an accessory pathway connecting the atrium and ventricle of a human heart109 and that successful surgical ablation of the accessory pathway results in the cure of the paroxysmal supraventricular tachycardia.110 The following three criteria developed by Mines for identification of circus movement reentry remains in use today:
An area of unidirectional block must exist.
The excitatory wave progresses along a distinct pathway, returning to its point of origin and then following the same path again.
Interruption of the reentrant circuit at any point along its path should terminate the circus movement.
It was recognized that successful reentry could occur only when the impulse was sufficiently delayed in an alternate pathway to allow for expiration of the refractory period in the tissue proximal to the site of unidirectional block. Both conduction velocity (CV) and refractoriness determine the success or failure of reentry, and the general rule is that the length of the circuit (path length) must exceed or equal that of the wavelength, the wavelength being defined as the product of the CV and the refractory period or that part of the path length occupied by the impulse and refractory to reexcitation. The theoretical minimum path length required for development of reentry was therefore dependent on both the CV and the refractory period. Reduction of CV111 or APD112 can both significantly reduce the theoretical limit of the path length required for the development of reentry.
That reentry could be initiated without the involvement of anatomic obstacles and that “natural rings are not essential for the maintenance of circus contractions“ was first suggested by Garrey in 1924.113 Nearly 50 years later, Allessie and coworkers4 provided direct evidence in support of this hypothesis in experiments in which they induced tachycardia in isolated preparations of rabbit left atria by applying properly timed premature extra stimuli. Using multiple intracellular electrodes, they showed that although the basic beats elicited by stimuli applied near the center of the tissue spread normally throughout the preparation, premature impulses propagate only in the direction of shorter refractory periods. An arc of block thus develops around which the impulse is able to circulate and reexcite its site of origin. Recordings near the center of the circus movement showed only subthreshold responses. The authors proposed the term leading circle to explain their observation.114 They argued that the functionally refractory region that develops at the vortex of the circulating wavefront prevents the centripetal waves from short circuiting the circus movement and thus serves to maintain the reentry. The authors also propose that the refractory core was maintained by centripetal wavelets that collide with each other. Because the head of the circulating wavefront usually travels on relatively refractory tissue, a fully excitable gap of tissue may not be present; unlike other forms of reentry, the leading circle model may not be readily influenced by extraneous impulses initiated in areas outside the reentrant circuit and thus may not be easily entrained. Although the leading circle reentry was widely accepted for a while as a mechanism of functional reentry, there is significant conceptual limitation to this model of reentry. For example, the centripetal wavelet was difficult to demonstrate either by experimental studies with high-resolution mapping or with computer simulation studies.
First introduced by Weiner and Rosenblueth in 1946,115 the concept of spiral waves (rotors) has attracted a great deal of interest over the past decade. Originally used to describe reentry around an anatomic obstacle, the term spiral wave reentry was later adopted to describe circulating waves in the absence of an anatomic obstacle.7,79 Because spiral waves of excitation is a well-described phenomenon in many excitable media,116 the application of the spiral waves of excitation to cardiac tissues is met with great enthusiasm. Spiral wave theory has advanced our understanding of the mechanisms responsible for the functional form of reentry. Although leading circle and spiral wave reentry are considered by some to be similar, a number of distinctions have been suggested. The curvature of the spiral wave is the key to the formation of the core.117 The curvature of the wave forms a region of high-impedance mismatch (source–sink mismatch), in which the current provided by the reentering wavefront (source) is insufficient to excite larger volume of tissue ahead (sink). A prominent curvature of the spiral wave is generally encountered after a wave break, where the wavefront meets the wavetail and a large curvature (and short action potential) is present. Because of a very small source in part related to a short action potential (where the wavefront and wavetail meet), the broken end of the wave moves most slowly. Figure 38–7 shows the formation of the spiral wave by wave front interaction with the refractory tail of a previous activation.118,119 This three-dimensional computer simulation study reproduced the wavebreak observed in the optical mapping studies of VF in swine ventricle. The wavebreak occurs when the wavefront encountered refractory tail of a previous activation, inducing two spiral waves (A). A three-dimensional view of the scroll wave is shown in B. C is an enlargement of the wavebreak. Note that the newly formed wavebreak has a very high curvature. As curvature decreases along the more distal parts of the spiral, propagation speed increases. The high curvature prevents the wave from propagating in the direction of wavebreak. The wavefront (red) then circles around the wavebreak site to form circus movement. In three dimensions, there are two new scroll waves formed by these interactions. Another difference between the leading circle and spiral wave is the state of the core; in former, the core is refractory because of repetitive centripetal wavelet that invades the core. In latter, the core remains unexcited because the source–sink mismatch prevented the propagation of the wavefront into the core.
Figure 38–7.

Three-dimension simulation of wavebreak by a wave front running into the trailing edge of refractoriness. A. Surface activation patterns (red = wave front; green = wave back) at the times indicated. The white arrows indicate the region where this mechanism of wave break occurs. B. Corresponding scroll wave fronts in the tissue (red = rising membrane voltage). C. Blowup of the region of wave break on the upper surface (near the whitearrows in A). Residual refractoriness (green) was left over by a previous wave front, and when the next wave (red) encountered this refractory region, wave break occurred, generating two new scroll waves. The white arrows point to the new wave breaks. At that site, the curvature of the wave is high, and the source-sink ratio is low, preventing the wave front from propagating. The wavefront (red) then circles around the wavebreak and forms circus movement. Reproduced with permission from Lee et al.119
The term spiral wave is usually used to describe reentrant activity in two dimensions. The center of the spiral wave is called the core, and the distribution of the core in three dimensions is referred to as the filament. The three-dimensional form of the spiral wave forms a scroll wave (see Fig. 38–7
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


