Congenital heart defects (CHDs) are the most prevalent of all birth malformations arising from the complex interplay of environmental exposures and genes. Modifiable environmental risk factors are still largely unknown, especially for paternal exposure. The aim of the present study was to examine the association between the environmental exposures of both parents and CHD risk and to explore the modification effect of metabolizing gene polymorphisms in children who lack the genetic capacity to produce the glutathione S-transferase (GST) GSTM1 and GSTT1 enzymes. A total of 330 parents of a child with CHD and 330 parents of a child without any congenital malformations were compared in terms of lifestyle habits and toxicant exposure. GST gene polymorphisms were investigated in 180 patients with CHD (104 males, age 4.9 ± 5.8 years). Paternal smoking (≥15 cigarettes/day) was significantly associated with CHD risk (odds ratio [OR] 2.1, 95% confidence interval [CI] 1.3 to 3.5, p = 0.002). Both maternal (OR 2.6, 95% CI 1.6 to 4.2, p <0.0001) and paternal (OR 2.5, 95% CI 1.6 to 3.8, p <0.0001) occupational/environmental exposures increased the risk of CHD. Also, a significant additive risk (OR 4.5, 95% CI 2.5 to 8.3, p <0.0001) was found when both parents were exposed to toxicants. Both maternal (OR 3.6, 95% CI 1.1 to 11.2, p = 0.03) and paternal (OR 3.3, 95% CI 1.0 to 10.8, p = 0.03) exposure to toxicants increased the CHD risk in children who carried the combined null GST genotypes. The effect for the combined null GST genotypes was also stronger (OR 6.5, 95% CI 1.5 to 28.0) when both parents were exposed. In conclusion, paternal smoking and exposure to toxicants for both parents affect the risk of children with CHD. Polymorphisms in GST genes can modify a person’s risk of toxicant exposure-induced disease.
Congenital heart defects (CHDs) are due to an abnormal development of the heart during embryogenesis and fetal life, between the second and ninth week of gestation. Generally, CHDs occur on a sporadic basis and do not have a familial history. Most CHDs are thought to have a multifactorial origin, with an interplay of genetic and environmental effect . It has recently been suggested that environmental risk factors could dominate in the etiology of CHD ; however, knowledge of such factors is very limited, especially for paternal exposure.
Furthermore, the effect of environmental factors might be modified by the genes responsible for the activation and detoxification of toxicant agents, contributing to an increased resistance (or sensitivity) to cardiac teratogenesis. In particular, metabolizing genetic polymorphisms of the glutathione S-transferase (GST) enzymes, which provide critical defense against numerous toxins, might modulate the effects of the toxic agent.
In the present study, we used a nested case-control design to examine the association between the environmental risk factors of both parents and CHD risk and to explore the modification effect of genetic susceptibility in children who lacked the genetic capacity to produce the GSTM1 and GSTT1 enzymes.
Methods
A prospective and paired case-control study (1:1) was conducted from October 2008 to April 2010 by inviting 360 parents of a child with isolated CHD and 360 parents of a child without any congenital malformations to participate. The study subjects were the parents of pediatric patients who had been admitted to our pediatric cardiac center and the parents of a healthy child who had been recruited in collaboration with the maternity and pediatric units. The matching criteria for the controls were the absence of a congenital malformation and age compatible with the observed age range of the patients. The exclusion criteria were mothers who reported inconsistent use of B vitamin and folate supplements in the periconception period; and an inability to obtain complete information about the occupational, demographic, and lifestyle data for both parents. The present study was conducted with informed consent of every participant, and the local ethical research committee approved it.
Both case and control parents completed a structured questionnaire on the demographic, preconceptional, and lifestyle exposures. We derived environmental and occupational exposure data from specific questions on potential teratogens/mutagens that have been linked to human reproductive impairment, including ionizing radiation, solvents, pesticides, asbestos, and heavy metals.
The paternal questionnaire carefully evaluated the 3-month period before conception, according to the duration of spermatogenesis (about 70 days). The maternal questionnaire covered the period before conception to the end of the first trimester of pregnancy. The first trimester of pregnancy was chosen because that it is most critical period for embryogenesis associated with increased sensitivity to environmental factors. The maternal questionnaire gathered information on personal characteristics and lifestyle habits, health, pregnancy aspects, medication during pregnancy, occupational, and other potential harmful exposures (e.g., diagnostic radiation exposure). The paternal questionnaire collected data on personal characteristics, health, occupational history, and other potential harmful exposures (e.g., diagnostic radiation exposure). Parental illness data included diseases of the thyroid, kidney, liver, gut, stomach, pancreas, gastrointestinal, respiratory, and urinary disorders.
Using a case-only design, DNA was obtained from 180 patients with CHD (104 males, age 4.9 ± 5.8 years). Genotyping analysis was performed by our laboratory staff, who were unaware of the clinical data. Genomic DNA was extracted from peripheral blood leukocytes. The GSTM1 and GSTT1 genotypes were determined using a co-amplification polymerase chain reaction approach with the GSTM4 gene, which is never deleted, as the internal control to distinguish the null genotypes from aborted polymerase chain reaction. Primer sequences, annealing temperatures, and digest conditions were performed according to previously published protocols. The internal standard fragment amplified from the GSTM4 gene was 157 bp. A 230-bp fragment was amplified for the GSTM1 gene, and a 480-bp fragment was obtained for the GSTT1 gene. The absence of amplified products was consistent with the null genotypes.
For statistical analysis, all determinants were dichotomized, except for the age variables. We considered as smokers those who smoked ≥3 cigarettes/day at conception; exsmokers were those who had stopped smoking ≥6 months before study inclusion; and nonsmokers were those who had never smoked. The smokers were also divided into groups according to smoking level: medium smokers (3 to 14 cigarettes/day) and heavy smokers (≥15cigarettes/day). The consumption frequency of alcoholic beverages (beer, wine, liquor) was classified in drinkers who drank ≥3 alcoholic drinks/day (beer, wine, liquor) and nondrinkers, who drank <3 drinks/day.
Given the relatively small number of exposed parents in any particular environmental and occupational category, statistical analysis for toxicant exposure was performed, including both environmental and occupational exposure to x-rays, chemicals, anesthetics, industrial cleaning agents and solvents, exhaust and welding fumes, paint/varnish/thinner, asbestos, heavy metals, and pesticides.
Statistical analyses of the data were conducted using the StatView statistical package, version 5.0.1 (Abacus Concepts, Berkeley, California). Data are expressed as the mean ± SD. Differences between the mean values of 2 continuous variables were evaluated using the unpaired Student t test. Differences in noncontinuous variables and genotype distribution were tested using chi-square analysis. Unconditional logistic regression analysis was used to estimate the odds ratios (ORs) and 95% confidence intervals (CIs) for the association between CHD and parental exposure. The ORs were also adjusted for potential confounding factors. A 2-tailed p value <0.05 was chosen as the level of significance. In the genetic study, we used a case-only study design because it is considered the most powerful method to study gene–gene and gene–environment interaction in disease etiology, achieving greater statistical power than a case-control study of the same size. With independence assumed between the exposure and the genotype, case-only ORs for the relevant interactions and 95% CIs were estimated by using the presence or absence of the GST variant as the dependent dichotomous variable in the logistic regression models that tested for an association with parental exposure. The study was powered to allow an interaction of magnitude of 2.3 with ≥80% power and an error rate of 5% under a sample size of 170 only-cases.
Results
The demographic characteristics of the study population are listed in Table 1 . The mothers of the patients with CHD had a significantly lower age than the controls (p = 0.01). No significant differences between the 2 groups were found for paternal characteristics. The CHD cases had a significantly lower birth weight (p = 0.01) compared to the control children. The children with CHD were also more often conceived with artificial fertilization (p = 0.05).
| Variable | Cases | Controls | p Value |
|---|---|---|---|
| Mother | |||
| Age (years) | 30 ± 5.4 | 31 ± 4.8 | 0.01 |
| History of chronic illness | 21 (12%) | 26 (14%) | 0.4 |
| Spontaneous miscarriage | 34 (19%) | 40 (22%) | 0.4 |
| Father | |||
| Age (years) | 33 ± 6.0 | 34 ± 5.1 | 0.3 |
| History of chronic illness | 29 (16%) | 33 (18%) | 0.6 |
| Offspring | |||
| Male gender | 104 (58%) | 105 (58%) | 0.9 |
| Birth weight (g) | 3,178 ± 630 | 3,340 ± 550 | 0.01 |
| Artificial fertilization | 6 (3.3%) | 1 (0.6%) | 0.05 |
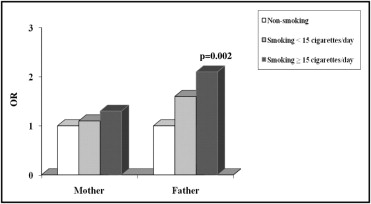
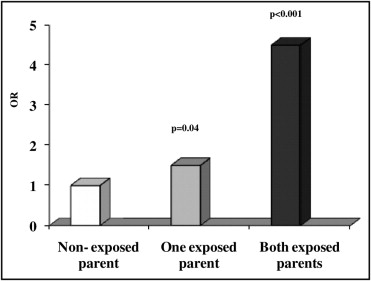
The parental exposure to environmental factors and CHD risk is listed in Table 2 . Maternal smoking was not significantly associated with an increased risk of CHD (OR 1.2, 95% CI 0.7 to 1.8). Fathers who were smokers showed an increased risk of CHD in their offspring (OR 1.7, 95% CI 1.1 to 2.6, p = 0.02), especially fathers who were heavy smokers (≥15 cigarettes/day) had a high risk of having children with CHD ( Figure 1 ) . Parental alcohol use did not influence the risk of CHD, and neither did the existence of a chronic disease or disease in the first trimester of pregnancy. Exposure to diagnostic radiographs was more frequently seen among the case-parents. Both maternal (OR 2.6, 95% CI 1.6 to 4.2, p <0.0001) and paternal (OR 2.5, 95% CI 1.6 to 3.8, p <0.0001) occupational/environmental exposure to toxicants increased the risk of CHD. The specific occupational and professional risk factors for CHD are listed in Table 3 . In addition, the exposure of both parents to toxicants showed a significant additive risk (OR 4.5, 95% CI 2.5 to 8.3, p <0.0001) of CHD in the offspring ( Figure 2 ) . On multivariate analysis, a father’s heavy smoking (OR 1.9, 95% CI 1.1 to 3.3) and maternal exposure (OR 2.1, 95% CI 1.2 to 3.6) and paternal exposure (OR 1.8, 95% CI 1.1 to 3.0) to toxicants were the main determinants of CHD risk. In the case-only study, we considered the association between GST polymorphisms in children and parental exposure. The frequency of GSTM1 null genotype was 57.7%, and the frequency of the GSTT1 null genotype was 16.7%. The genotype distribution was within the range previously reported for GSTM1 and GSTT1 null genotypes in other white populations. We did not detect an interactive effect of GSTM1 or GSTT1 null genotypes and active maternal and paternal smoking ( Table 4 ). The risk of CHD tended to be greater in children carrying the GSTM1 and GSTT1 null genotypes who had parents exposed to toxicants. A significant interaction was found for the combined null GSTs and both maternal (OR 3.6, 95% CI 1.1 to 11.2, p = 0.03) and paternal (OR 3.3, 95% CI 1.0 to 10.8, p = 0.03) exposure to toxicants ( Figure 3 ) . Finally, children with the combined null GST genotypes had a greater risk (OR 6.5; 95% CI 1.5 to 28.0) than children carrying wild-type GST genes when both parents were exposed ( Figure 3 ).
| Variable | CHD Parents (n = 360) | Control Parents (n = 360) | OR (95% CI) | p Value |
|---|---|---|---|---|
| Active smoking (positive) | ||||
| Mother | 50 (27.8%) | 45 (25.0%) | 1.2 (0.7–1.8) | 0.6 |
| Father | 81 (45.0%) | 65 (36.1%) | 1.7 (1.1–2.6) | 0.02 |
| Diagnostic x-ray exposure (positive) | ||||
| Mother | 49 (27.2%) | 38 (21.1%) | 1.3 (0.8–2.2) | 0.2 |
| Father | 45 (25.0%) | 37 (20.6%) | 1.3 (0.8–2.1) | 0.3 |
| Heavy drinking (positive) | ||||
| Mother | 1 (0.6%) | 1 (0.6%) | 1.0 (0.1–16) | 0.99 |
| Father | 7 (3.9%) | 7 (3.9%) | 1.0 (0.3–2.9) | 0.99 |
| Occupational/environmental exposure (positive) | ||||
| Mother | 70 (38.9%) | 34 (18.9%) | 2.6 (1.6–4.2) | <0.0001 |
| Father | 88 (48.9%) | 49 (27.2%) | 2.5 (1.6–3.8) | <0.0001 |
| Variable | CHD Parents | Control Parents | p Value |
|---|---|---|---|
| Maternal occupational exposure | 0.004 | ||
| Risk factor (n) | 28 (15.5%) | 11 (6.1%) | |
| Paint/varnish/thinner/solvents | 9 | 0 | |
| X-rays (hospital workers) | 4 | 2 | |
| Cleaning agents and solvents | 4 | 1 | |
| Industrial chemicals and solvents | 4 | 1 | |
| Hair dye | 2 | 2 | |
| Textile dye | 2 | 0 | |
| Chemical laboratory exposure | 1 | 1 | |
| Asbestos | 1 | 0 | |
| Pesticides | 1 | 0 | |
| Anesthetic gas | 0 | 4 | |
| Paternal occupational exposure | 0.003 | ||
| Risk factor (n) | 60 (33.3%) | 41 (22.8%) | |
| Paint/varnish/thinner/solvents (shoe factory) | 14 | 4 | |
| Cement dust | 15 | 8 | |
| Industrial chemicals and solvents | 11 | 6 | |
| Hair dyes | 1 | 1 | |
| Cleaning agents and solvents | 1 | 1 | |
| X-rays (hospital workers) | 1 | 4 | |
| Textile dye | 1 | 0 | |
| Exhaust and welding fumes | 2 | 3 | |
| Chemical laboratory exposure | 1 | 0 | |
| Asbestos | 6 | 1 | |
| Heavy metals | 4 | 5 | |
| Pesticides | 3 | 1 | |
| Anesthetic gas | 0 | 7 | |
| Maternal environmental exposures | 0.01 | ||
| Risk factor (n) | 45 (25.0%) | 25 (13.9%) | |
| Waste sites | 9 | 0 | |
| Thermal power plant | 8 | 1 | |
| Industrial pollution | 9 | 5 | |
| Electromagnetic field | 4 | 0 | |
| Environmental pollution | 3 | 3 | |
| X-rays (radiotherapy; Chernobyl) | 2 | 0 | |
| Asbestos | 1 | 1 | |
| Pesticide sites | 5 | 0 | |
| Drugs | 4 | 15 | |
| Paternal environmental exposure | <0.0001 | ||
| Risk factor (n) | 40 (22.2%) | 12 (6.7%) | |
| Waste sites | 9 | 0 | |
| Thermal power plant | 9 | 0 | |
| Industrial pollution | 8 | 5 | |
| Electromagnetic field | 3 | 0 | |
| Environmental pollution | 3 | 0 | |
| Asbestos | 0 | 1 | |
| Pesticide sites | 5 | 0 | |
| Drugs | 3 | 6 |
| Positive Parental Exposure | GSTM1 | OR (CI 95%) | p Value | |
|---|---|---|---|---|
| Present | Absent | |||
| Mother | ||||
| Active smoking | 0.3 | |||
| Negative | 54 | 61 | 1.0 | |
| Positive | 19 | 31 | 1.4 (0.7–2.9) | |
| Diagnostic x-ray exposure | 0.1 | |||
| Negative | 61 | 70 | 1.0 | |
| Positive | 17 | 32 | 1.6 (0.8–3.2) | |
| Other toxicant exposures | 0.2 | |||
| Negative | 52 | 58 | 1.0 | |
| Positive | 26 | 44 | 1.5 (0.8–2.9) | |
| Father | ||||
| Active smoking | 0.8 | |||
| Negative | 34 | 45 | 1.0 | |
| Positive | 36 | 45 | 1.0 (0.5–1.8) | |
| Diagnostic x-ray exposure | 0.6 | |||
| Negative | 60 | 75 | 1.0 | |
| Positive | 18 | 27 | 1.2 (0.6–2.4) | |
| Other toxicant exposures | 0.1 | |||
| Negative | 45 | 47 | 1.0 | |
| Positive | 33 | 55 | 1.6 (0.9–2.9) | |
| GSTT1 | ||||
| Mother | ||||
| Active smoking | 0.1 | |||
| Negative | 93 | 22 | 1.0 | |
| Positive | 45 | 5 | 0.5 (0.2–1.3) | |
| Diagnostic x-ray exposure | 0.9 | |||
| Negative | 109 | 22 | 1.0 | |
| Positive | 41 | 8 | 1.0 (0.4–2.3) | |
| Other toxicant exposures | 0.08 | |||
| Negative | 96 | 14 | 1.0 | |
| Positive | 54 | 16 | 2.0 (0.9–4.5) | |
| Father | ||||
| Active smoking | 0.4 | |||
| Negative | 63 | 16 | 1.0 | |
| Positive | 69 | 12 | 0.7 (0.3–1.4) | |
| Diagnostic x-ray exposure | 0.1 | |||
| Negative | 109 | 26 | 1.0 | |
| Positive | 26 | 4 | 0.4 (0.1–1.2) | |
| Other toxicant exposures | 0.09 | |||
| Negative | 81 | 11 | 1.0 | |
| Positive | 69 | 19 | 2.0 (0.9–4.5) | |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


