Prevalence and epidemiology
A number of epidemiologic studies are now available showing that HCM occurs in at least 0.2% of the general population (e.g. 1:500).8,19–22 This prevalence is greater than previously regarded, although HCM remains relatively uncommon in general cardiology practice.23 This paradox strongly suggests that most individuals affected by HCM do not achieve clinical recognition. Furthermore, there is growing evidence that HCM is truly a global disease,20 although to date the most intense interest and reporting of cases has been in North America, Western Europe and Asia (Japan and China). HCM occurs equally in the genders and has been reported in many racial groups, including African-Americans.24
HCM is characterized by a thickened but non-dilated left ventricle (LV) in the absence of another cardiac or systemic disease capable of producing the magnitude of hypertrophy evident (e.g. aortic valve stenosis, systemic hypertension, and some phenotypic expressions of athlete’s heart).2–8 For the past 35 years, the clinical diagnosis of HCM has usually been made by echocardiographic imaging.3 Cardiovascular magnetic resonance (CMR) has an expanding role in the non-invasive diagnosis of HCM. For example, CMR may visualize areas of segmental hypertrophy, specifically in the anterolateral free wall and LV apex, not reliably identified with echocardiography.25
Hypertrophy
The distribution of LV hypertrophy is characteristically asymmetric, in which some portion of the LV wall is thicker than other areas.7 Frequently, the pattern of hypertrophy is strikingly heterogeneous, with contiguous segments of LV wall differing greatly in thickness. Hypertrophy may be diffuse, involving substantial portions of ventricular septum and LV free wall, including patients with the most substantial hypertrophy observed in any cardiac disease.7 However, in about one-third of patients, LV hypertrophy may be relatively mild and confined to small areas of the chamber such as basal anterior septum or apex. Furthermore, genotype-phenotype studies in HCM families have documented the association of HCM mutant genes with near-normal or even normal LV wall thicknesses.12,26 Therefore, virtually any LV wall thickness is compatible with the clinical and/or genetic diagnosis of HCM.
Histopathology
Cardiac muscle cells (myocytes) in ventricular septum and LV free wall have increased transverse diameter as well as bizarre shapes, often maintaining intercellular connections with several adjacent cells.27,28 Frequently, these myocytes are arranged in a chaotic, disorganized pattern at oblique and perpendicular angles to each other; myofibrils within cardiac muscle cells may also be in disarray. Disorganized cardiac muscle cells occur in about 95% of patients dying of HCM and usually occupy substantial portions of the LV, about 33% of septum and 25% of free wall.27,28
Microvascular abnormalities of intramural coronary arteries are present in about 80% of patients studied at necropsy, most commonly in the ventricular septum.29 These vessels commonly have thickened arterial walls due to increased intimal and/or medial components, associated with apparent luminal narrowing. This form of “small vessel disease” may be responsible for regional myocardial ischemia,11 necrosis, and ultimately replacement fibrosis with grossly visible extensive (or even transmural) scars. It is likely that disorganized myocardial cells and architecture, as well as areas of replacement fibrosis dispersed throughout the LV wall, impair normal transmission of electrophysiologic impulses, predispose to disordered patterns and increased dispersion of electrical depolarization and repolarization, and probably serve as an electrically unstable arrhythmogenic substrate with the potential to trigger lethal ventricular tachyarrhythmias and sudden death.30 Myocardial disorganization and fibrosis could also contribute to diastolic dysfunction and progression of heart failure.
LV outflow obstruction
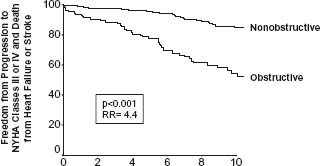
Dynamic subaortic obstruction in HCM is usually produced by systolic anterior motion (SAM) of the mitral valve, resulting in contact between mitral valve and ventricular septum.6 Subaortic obstruction in HCM may fluctuate in response to a number of provocations (including exercise)31,32 but represents true mechanical impedance to LV outflow. The markedly increased intraventricular pressures that result may be detrimental to LV function by increasing myocardial wall stress and oxygen demand. LV outflow tract obstruction (gradient ≥30mmHg) is a proven determinant of progressive heart failure symptoms and cardiovascular death over many years9 (9) (Fig. 49.1). Furthermore, fully 70% of an HCM cohort has the propensity to develop LV outflow obstruction either at rest or with physiologic exercise.10
Figure 49.1 Long-term significance of LV outflow tract obstruction. Probability of progression to severe heart failure (NYHA III or IV) or death from heart failure or stroke among 224 patients with LV outflow tract obstruction (gradient at rest ≥ 30 mmHg), and 770 patients without obstruction. (From Maron et al9 with permission of the Massachusetts Medical Society.)
HCM is a Mendelian trait with an autosomal dominant pattern of inheritance. Molecular studies with clinical genotype-phenotype correlations have convincingly demonstrated that HCM is caused by mutations in any one of 11 genes, each encoding a protein component of the cardiac sarcomere (of either the thick or thin filaments) with contractile, structural or regulatory functions.12,26,33–38
Two of the HCM-causing mutant genes, beta-myosin heavy chain and myosin-binding protein C, predominate in frequency. The other nine genes account for far fewer cases of HCM and include troponin T and I, regulatory and essential myosin light chains, titin, alpha-tropomyosin, alpha-actin, alpha-myosin heavy chain and muscle LIM protein (MLP). This genetic diversity is compounded by the considerable intragenic heterogeneity with over 400 mutations now identified (http://cardiogenomics.med. harvard.edu). The characteristic morphologic diversity of the HCM phenotype is largely attributable to the disease-causing mutations, but probably also to the influence of modifier genes and environmental factors on phenotypic expression.
Onset of heart failure symptoms often occurs in early adulthood between 20 and 40 years of age, although symptoms can become evident at any age, from young children to the elderly.3,39,40 While some degree of heart failure is frequent in HCM, progression to severe functional limitation (i.e. NYHA class III–IV) is uncommon, occurring in about 10–15% of hospital-based patient populations.3,4 The usual circumstance is characterized by exertional dyspnea and fatigue (and sometimes orthopnea and paroxysmal nocturnal dyspnea), indicative of elevated pulmonary venous pressures in the presence of intact or hyperdynamic systolic function. Such symptoms are related to diastolic dysfunction, outflow tract obstruction, microvascular myocardial ischemia, or a combination of these pathophysiologic variables. HCM patients may also experience syncope (or near-syncope), lightheadedness, palpitations, and chest pain, either typical of midsternal angina pectoris or with more atypical features.
Overall patient population
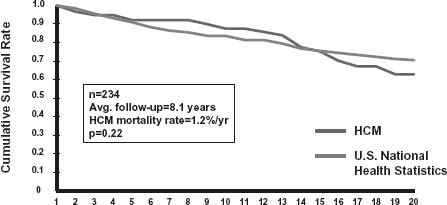
Predicting clinical course and outcome for individual HCM patients continues to be challenging because of the marked variability and heterogeneity in disease expression, particularly for young patients with their long period of potential risk. Annual premature HCM-related mortality rates were initially reported to be 3–6% with the higher range in children.5,41 However, these are now regarded as outdated estimates derived largely from highly selected HCM cohorts, skewed toward high-risk patients at tertiary referral centers. Greater insight into the clinical course of HCM has been achieved by analysis of unselected community-based patient populations, uncontaminated by tertiary center referral bias. These cohorts have allowed more realistic overall mortality rates of about 1%/year to be established, although somewhat higher for children (i.e. 2%/ year)3,40,41 (Fig. 49.2). Therefore, in adults, HCM does not add to total mortality over that expected for the general population and is compatible with normal life expectancy, often without significant disability or the necessity for major interventions.3,39,40
Figure 49.2 Cumulative survival in a community-based adult HCM population. Total mortality in 234 HCM patients (1%/ year) does not differ significantly from that expected in general US population after adjustment for age, sex, and race. (From Maron et al,40 reproduced with permission of the American Medical Association.)
Risk stratification and sudden cardiac death
Demographics
Sudden death in HCM may occur at a wide range of ages, but most commonly during adolescence and young adulthood, less than age 30-35 years of age.3-6,41–45 Indeed, HCM is the most common cause of sudden cardiac death in young people and these events are arrhythmia-based due to primary ventricular tachycardia/fibrillation, with a predilection for the early morning hours.30,45,46 Sudden death is often the initial disease manifestation in asymptomatic individuals, many of whom are undiagnosed during life. Although most patients die suddenly while sedentary or during normal/modest physical activity, an important proportion do so associated with vigorous exertion, including young competitive athletes in organized sports. This observation is the basis for the standard and prudent recommendation to disqualify young athletes with HCM from intense competitive sports to reduce their risk for sudden death, according to the guidelines of Bethesda Conference #36.47
Risk factors3–5,30,43,44,48–54
For secondary prevention, risk for sudden death is associated with prior cardiac arrest or sustained ventricular tachycardia. For primary prevention, one or more of the following clinical risk markers are relevant:
- family history of ≥1 premature HCM-related deaths, particularly if sudden and multiple
- unexplained recent syncope, particularly in the young and related to exertion
- hypotensive or attenuated blood pressure response to exercise
- multiple repetitive (or prolonged) non-sustained ventricular tachycardia on serial Holter (ambulatory) ECG monitoring
- massive LV hypertrophy (wall thickness ≥30 mm), most relevant to younger patients.
Each of these risk factors, taken individually, has potential limitations and all are encumbered with relatively low positive predictive value (of about 20%), although with high negative predictive accuracy. For example, the finding of non-sustained ventricular tachycardia (NSVT) on ambulatory Holter may be fraught with particular ambiguity48,53,54 A single brief isolated burst of NSVT on a random Holter ECG is no longer regarded as a risk factor which itself would trigger a primary prevention implanted cardioverter-defibrillator (ICD). However, long runs of NSVT (>10 beats) intuitively carry greater weight in these clinical circumstances. One potentially useful (but non-evidence based) strategy utilized following identification of a short run of NSVT on the initial Holter ECG involves expanding the monitoring period to a total of six days by obtaining additional ECG recordings at about 1-2 week intervals.53,54 This approach allows assembly of an extended profile of ectopy Thus, a more measured assessment of an individual patient’s day-to-day ventricular ectopy (including NSVT) is achieved, potentially clarifying the decision concerning a prophylactic ICD.
It has been proposed that certain mutations responsible for HCM could represent prognostic markers conveying either favorable or adverse outcome, including the risk for sudden death.12,55 For example, early genotype-phenotype studies identified certain beta-myosin heavy chain and troponin T mutations to be associated with higher frequency of premature death compared to other mutations, such as those involving myosin-binding protein C or alpha-tropo-myosin. More recently, however, the prognostic significance of disease-causing mutations for risk stratification and decision-making concerning treatment strategies has been questioned and de-emphasized.37,38 Due to the substantial genetic heterogeneity in HCM and lack of compelling data, it is now evident that anticipating the future clinical course based on knowledge of the disease-causing mutation is untenable and has been largely abandoned in the management of individual patients.
While the presence of a subaortic gradient (≥30mmHg at rest) is a strong determinant of progressive heart failure and cardiovascular death, the relationship is much less predictive of sudden cardiac death9 and LV outflow obstruction should not be regarded as a primary risk marker.
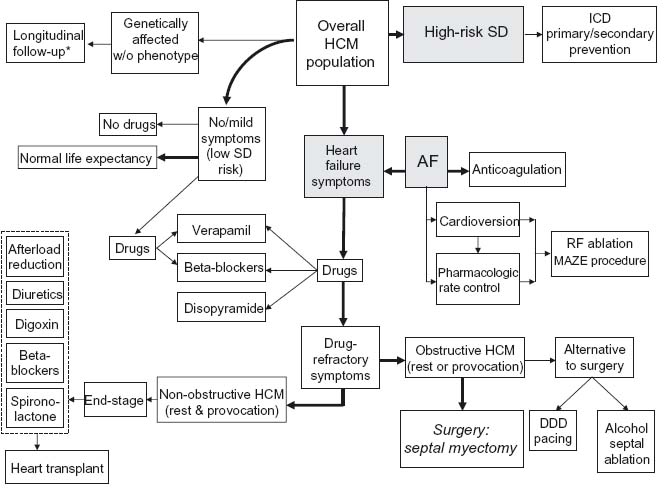
Treatment strategies in individual HCM patients are usually tailored along one of three major prognostic pathways: disabling symptoms due to heart failure, high risk for sudden death, and atrial fibrillation (Fig. 49.3).
Figure 49.3 Current treatment strategies for patient subgroups within HCM disease spectrum. Management decisions usually proceed along three major prognostic pathways: disabling symptoms due to heart failure, high risk for sudden death, and atrial fibrillation. * No specific treatment or intervention indicated, except under exceptional circumstances. A F, atrial fibrillation; ICD, implantable cardioverter-defibrillator; R F, radiofrequency; SD, sudden death.
Medical management of heart failure
Data dictating pharmacologic treatment of symptomatic HCM patients with heart failure are largely derived from clinical experience in selected patient subsets followed for relatively brief periods of time,56 with the exception of one double-blind cross-over study in a small patient cohort.15 When associated with normal or hyperdynamic LV function the response of heart failure symptoms to medical treatment is highly variable; hence, therapy is often tailored empirically to the individual requirements of symptomatic patients. Since the mid-1960s a variety of beta-adrenergic receptor-blocking drugs have been utilized extensively to relieve and control heart failure symptoms in patients with obstructive or non-obstructive HCM.1 Verapamil may improve cardiac symptoms and exercise capacity (largely in non-obstructive patients) due to a beneficial effect on LV relaxation and filling, but should be avoided in the presence of elevated pulmonary venous pressures and marked outflow obstruction.3 In patients who fail to benefit from beta-blockers and verapamil, an empiric trial may be initiated with disopyramide (in combination with a beta-blocker).57
Alternatively, about 3% of HCM patients will manifest the “end-stage” of HCM in which progressive heart failure is associated with LV systolic dysfunction and often remodeling in the form of wall thinning and cavity dilation.58 Drug therapy in these patients is similar to that employed for congestive heart failure in other cardiac diseases and may include the administration of beta-blockers, ACE inhibitors, angiotensin receptor blockers and diuretics, as well as possibly digoxin, spironolactone, warfarin, and ultimately heart transplant.
Prevention of sudden death
Historically, the management of high-risk HCM patients was first confined to prophylactic pharmacologic treatment with beta-blockers, verapamil, and antiarrhythmic agents (such as procainamide, quinidine, and subsequently amiodarone). However, in HCM there are virtually no data supporting the efficacy of prophylactic drug treatment in preventing sudden death.59
When the level of risk for sudden death is judged by risk factor analysis to be unacceptably high and deserving of intervention, the ICD has proved to be the most effective available prophylactic treatment option, with the potential for absolute protection and alteration in the natural history of this disease by aborting potentially life-threatening ventricular tachyarrhythmias.30,46,60 Unavoidably, all available ICD data in HCM are observational and by design non-randomized, with patients accessed retrospectively and prospectively.61–63
The largest such study cohort is composed of 506 HCM patients in an international multicenter registry, all of whom had ICDs implanted for high-risk status, and were followed for an average period of about 3½ years (up to 16).30 Appropriate device discharges (either defibrillation shocks or antitachycardia pacing) triggered by ventricular tachycardia/fibrillation occurred in 20% of patients with an overall annual discharge rate of 5.5%. Appropriate intervention rate was 11%/year for secondary and 4%/year for primary prevention. Of note, almost 30% of the young patients implanted at ≤ 20 years of age subsequently had appropriate ICD discharges at 18 ± 4 years of age. Therefore, the ICD has proven effective in HCM despite the often substantially increased cardiac mass characteristic of this disease, as well as the not infrequent occurrence of LV outflow tract obstruction. Furthermore, about 40% of those patients who received appropriate defibrillator therapy to terminate potentially lethal ventricular tachyarrhythmias in fact experienced multiple such interventions.
Medical treatment is not absolutely protective against the risk of sudden death in HCM. Indeed, in one large study, a substantial proportion of patients were taking amiodarone or other potentially antiarrhythmic drugs at the time of appropriate defibrillator interventions.59
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


