Lymphocyte- and Macrophage-Mediated Inflammation in the Lung
INTRODUCTION
The lung receives a continual flow of foreign infectious and noninfectious antigens during respiration. Like the gut, genitourinary tract, and skin the lung is one of the interfaces of the sterile body with the environment. The lung’s immune defense system and inflammatory mechanisms are poised to deal with this role. In this chapter we will consider the inflammatory and immune roles of two key cells of hematopoietic origin: the lung lymphocyte and the lung macrophage.
While these two cell types interact extensively and might even be considered co-dependent in many situations, they represent two very different arms of the inflammatory response. The macrophage, as a phagocytic cell, is of ancient phylogenetic lineage. It is a sentinel of the innate immune system. As such, it is not antigen specific but it is triggered by many inflammatory stimuli through both specific and pattern recognition receptors. Lymphocytes are present only in vertebrates and represent a significant refinement in the inflammatory response by the ability to recognize and adapt to specific antigens and discriminate between self and nonself. The functional distinction between these two arms of the immune system is blurring with the recent discovery of innate lymphoid-derived immune cells and the appreciation of multiple functions of macrophages in the inflammatory response.
Macrophages or dendritic cells (DCs) are required for optimal presentation of antigens to lymphocytes, and for optimal lymphocyte activation and cytokine production. Conversely macrophage microbicidal function and release of arachidonate and oxygen metabolites is influenced by cytokines produced by activated T lymphocytes and phagocytosis is markedly enhanced by antibodies produced by B lymphocytes. The cooperation between these two cell types represents a cornerstone of lung defense against noninfectious antigen challenge or microbial infection. Another chapter will deal with acute lung inflammation mediated by neutrophilic leukocytes. In this chapter we will present a brief overview of the macrophage and lymphocytes in the human lung, their function and interactions, and a synthesis of their role in lung inflammation and disease.
We will assume a basic knowledge of immunology. However, an explanation of the terminology used in this chapter is appropriate. Many surface receptors expressed by immunologic cells have had multiple names based on different functions. In the past 30 years these terms have been grouped together in a series of standardized “clusters of differentiation” (CD) for the purpose of standard nomenclature. A list of the CD markers referred to in this chapter, other names used for them, and their putative functions are included in Table 21-1.
TABLE 21-1 Cluster of Differentiation Antigens and Surface Molecules Discussed in this Chapter

LYMPHOCYTES IN THE LUNG
Lymphocytes are much more abundant in the lung than previously documented. Recent estimates from CD3+ stained cells in histologic sections of normal human lung approximate the resident T-cell pool to be 10 billion T cells, which is comparable to the number of T cells in human blood.1 These resident T cells have been examined in detail (see2) and are primarily memory T cells with a resident effector phenotype (rTem). In the normal lung these lymphocytes are distributed in one of four compartments (Fig. 21-1). The compartments include lymphocytes at the epithelial surface (LES) including those in the bronchoalveolar space; lymphocytes associated with the epithelium in lymphoid aggregates (also known as bronchus-associated lymphoid tissue [BALT]); interstitial and intraepithelial lymphocytes (IELs); and an intravascular pool. Although the presence of BALT in normal human lung is controversial, it is clear that BALT is present in the setting of infection and possibly with chronic airway inflammation.3,4 Each compartment has a distinct phenotypic and functional repertoire. It is not yet clear whether there is a sequential influx of lymphocytes from the blood/intravascular pool to interstitium or BALT and finally to the epithelial surface, or whether lymphocytes are destined to reside in one or another of these pools from the time of maturation and/or activation. The absence of afferent lymphatics to the lung dictates that the intravascular pool is the original source of lymphocytes destined to one of the pulmonary compartments. The exact nature of influx and turnover of normal lung lymphocyte populations is not clear. However, the identification of organ- and lung-specific homing chemotactic cytokines (chemokines) with selective distribution of their cognate receptors raises the possibility that the origins of the populations of each compartment are distinct and for intercompartmental trafficking to occur, there must be site-specific signals that alter the chemotactic receptor repertoire in situ.
 LYMPHOCYTES AT THE EPITHELIAL SURFACE (LES)
LYMPHOCYTES AT THE EPITHELIAL SURFACE (LES)
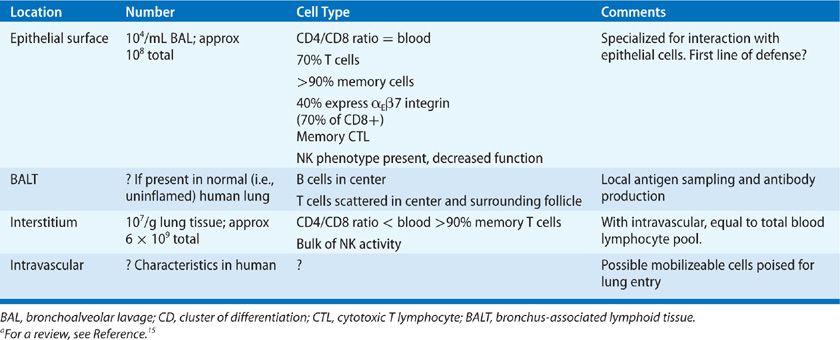
LES and IELs are best studied as lymphocytes from the bronchoalveolar space, are easily recovered from the lung using bronchoalveolar lavage (BAL). Although in normal nonsmoking individuals lymphocytes make up only about 5% to 15% of the 105 cells found per milliliter of BAL fluid, lymphocyte numbers may increase dramatically during an inflammatory response.5–7 LES differ markedly from blood lymphocytes suggesting either a selection bias or organ-specific maturation process that occurs between the blood and lung (Table 21-2). Approximately 70% of LES are T cells; the CD4/CD8 ratio of T cells is approximately the same as in the blood, though with a larger scatter among individuals.8 Over 70% of BAL T cells are of the previously activated memory type as determined by expression of a low–molecular-weight form of the leukocyte common antigen CD45 (CD45RO),9,10 and many have been chronically activated as shown by expression of the α1β1 integrin. The balance of BAL T cells are naive, based on their expression of one of the chemokine receptors CCR7,11 which primarily identifies T cells that have not come into contact with their cognate antigen. It is unclear what the T-cell representation in BAL is of IEL compared to LES. LES are more likely than blood T cells to express the activation antigen HLA-DR, and CD8+ (cytotoxic/suppressor) and LES are more likely to express markers associated with cytotoxic cell function.5 An unusual population of memory cytotoxic cells which lack the accessory molecule CD28 has also been described in normal LES.12 In the neonatal rodent lung the majority of T cells express the gamma-delta T-cell receptor, and account for about 20% of resident (whole lung) pulmonary T cells in adult mice—a percentage that is markedly upregulated with infection.13 By contrast, the vast majority of human T cells at the epithelial surface are alpha-beta+. The function of the small population of gamma-delta LES T cells in humans remains unknown, but functional similarities to homologous mouse cells suggest that their primary role is to regulate the primary immune response.14 There are a variable number of natural killer (NK) (including NK-T) cells in this compartment as might be expected in an area of microbial and antigen assault. However, the bulk of NK activity in the lung is found in the interstitial population.15,16 B cells are also present in the LES population derived by BAL. They have been documented to produce antibodies of all types, with their primary role being to provide mucosal immunity through the secretion of IgA.5 It is unknown whether there are selected B cell populations among LES.
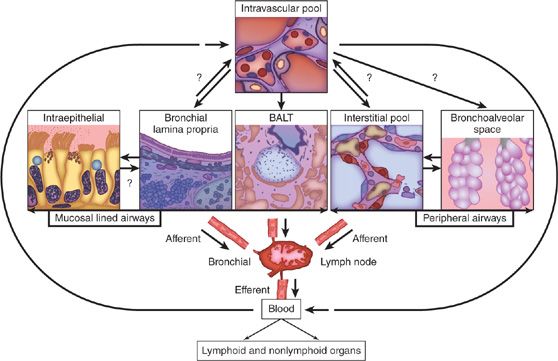
Figure 21-1 Lymphocytes are found in the lung in distinct sites. These include LES (including those at the bronchoalveolar surface); the interstitial and intraepithelial lymphocytes (IELs), bronchus-associated lymphoid tissue (BALT) which are centers of airway antibody production; and an intravascular pool. Lymphocytes travel from lymph nodes to blood and into lung via interaction with lung endothelial cells. Arrows indicate hypothesized trafficking from one pulmonary compartment to another. Some lymphocytes from these various compartments may be able to exit the lung back to lymph nodes, and others become effete and die. For further explanation see text. (Reproduced with permission from Pabst R. IS BALAT a major component of the human lung immune system? Immunology Today. 1992;13(4):119–122.)
The source and fate of these T and B cells are unknown. It seems reasonable to hypothesize that epithelial surface T cells (and other lung T cells) emerge from the circulation, perhaps proliferate locally and differentiate further while in the lung, and then die or recirculate.17 The appearance of labeled blood T cells in LES have been found in the circulation. The predominant memory phenotype of epithelial surface and interstitial T cells strongly suggests that such differentiation occurs before entry into the lung. Once in the epithelial compartment it appears that bronchial IEL T cells can be long lived potentially surviving within the epithelium for several months, in contrast to the short life span of lymphocytes in the lamina propria.18,19
TABLE 21-2 Characteristics of Lung Lymphocytesa
Many LES adhere to and interact with airway epithelial cells through the expression of a unique adhesion molecule (HML-1/αeβ7 integrin).20,21 This integrin is expressed on 40% of LES (60% of CD8+ cells are HML-1+ while fewer CD4+ cells express it) and on intestinal lymphocytes, but only rarely on blood or lung interstitial lymphocytes. It is likely that local influences, such as epithelial-derived cytokines like transforming growth factor beta-1 (TGFβ1), result in the expression of this molecule on LES.22–24 Epithelial cells are directly stimulated by bacteria to release specific chemokines and cytokines (e.g., interleukin (IL)-8, MIP2α (CXCL2), MIP3α (CCL20), IL-7, IL-15) depending on the organism and pathogenicity.25 These ligands can bind surface chemokine and cytokine receptors on adjacent LES, suggesting that these cells are not necessarily effete or dying cells, which are present in the airway only to be cleared by the mucociliary escalator and expectorated. Rather, they include a specialized lymphocyte population involved in the surveillance of the airway and interaction with epithelial cells. The possibility that LES re-enter the interstitium and lymphoid tissue has been confirmed experimentally in rats.26 In addition to their interaction with airway epithelial cells, LES directly interact with mucosal DCs, whose phenotype directs further T-cell phenotype evolution (see Lymphocyte Activation in the Lung, below)
LES can be stimulated to proliferate, produce cytokines and antibodies, and to perform cytolytic functions. However, they are in general hyporeactive in proliferative or antibody responses to antigen or mitogens when compared to blood T cells or even when compared to memory T cells in lung interstitium. The reason for this is not known, but may relate to immunosuppressive influences in the airways, including alveolar macrophages (AMs),27 local production of TGFβ1,28 the immunosuppressive activity of pulmonary surfactant lipids or proteins,29 and the possible presence of other immunomodulatory cytokines like IL-10 and IL-16.
 BRONCHUS-ASSOCIATED LYMPHOID TISSUE (BALT)
BRONCHUS-ASSOCIATED LYMPHOID TISSUE (BALT)
BALT is the term applied to localized subepithelial collections of lymphocytes in the airways, and is a secondary lymphoid tissue analogous to other types of mucosa-associated lymphoid tissue (reviewed by Berman et al.,6 in-depth review by Randall30). BALT is present in normal rodent airway and increases in amount with age. Current evidence suggests these structures are uncommon or absent in adult humans but are present in childhood and may appear and proliferate later in life in response to infection or chronic inflammation.3,30–35 These data suggest that inflammation or infection induces the development of these lymphoid aggregates in humans, and are termed inducible BALT (iBALT) to distinguish them from the classically defined BALT observed in rodents, which is believed to be formed independent of antigen.36
Classically defined BALT is similar to gut-associated lymphoid tissue (GALT; e.g., Peyer’s patches) in appearance, association with epithelium and a blood vessel, presence of specialized cuboidal or high endothelial venules characteristic of lymphoid tissues, and a specialized thinned overlying epithelium facilitating antigen entry from the bronchial lumen and exit of lymphocytes and lymphocyte products. Immunohistochemistry has revealed a preponderance of B cells staining with IgM, IgG, and IgA, with a scattering (approximately 20%) of T cells, especially CD4+ helper cells within and surrounding the aggregate. BALT lacks organized germinal centers found in other secondary lymphoid tissue. The resemblance of BALT to GALT, as well as the similarity of lymphocyte recirculation patterns from lung- and gut-associated lymphoid tissue has suggested to some authors that these structures represent a common mucosal immune system. In this paradigm, recirculating blood lymphocytes exit into these structures which provide an efficient exposure to antigens sampled from the environment. Activated memory cells are then a source of local antibody production, and they may disperse through the circulation to other mucosal sites to provide dissemination of immunologic memory.
Despite these observations that have been made in rodents, several issues continued to limit what we can infer about BALT function in humans. They include its near absence in normal human airways and limited presence in experimental animals which are primarily observed in pathogen-free colonies. In fact, the infrequent appearance of BALT in humans and mice has led some to doubt whether BALT is an important secondary lymphoid organ.31 However, there is accumulating evidence that iBALT plays a role in the adaptive immune response to infection.36 In this new paradigm, infection and inflammation trigger the development of these localized lymphoid tissues through recruitment and priming of naive lymphocytes thereby generating antigen-specific lymphocytes in situ. At the same time, infection and inflammation activate local antigen-presenting cells, which migrate to conventional secondary lymphoid organs (i.e., lymph nodes) for priming and activation of effector cells. Thus, these local lymphoid tissues are capable of not only expanding effector cells that were primed in conventional secondary lymphoid organs but also initiating primary immune responses in situ. In support of this paradigm, splenectomized lymphotoxin-α-null mice that lack the ability to form any secondary lymphoid tissues are still able to develop iBALT36 in response to infection suggesting distinct mechanisms govern over these lymphoid tissues.
The uniqueness of iBALT in humans and mice is highlighted by the structural difference compared to constitutive BALT in rodents. The iBALT can vary in organization from small clusters of B cells, T cells, and DCs to well-developed follicular aggregates and unlike constitutive BALT, which is found in the upper airway of rodents, iBALT is found in perivascular, peribronchial, and even interstitial areas in the lower airways of the lung.36 In humans, the specialized overlying epithelium has not been characterized.37 B-cell follicles, which are seen in iBALT, are centered around CD21-expressing follicular dendritic cells (FDCs)36 and separated by interfollicular regions containing resident CD11c+ DCs and both CD4+ and CD8+ T cells.38 The lymphocyte homing chemokines CXCL13 and CCL21 are required for recruitment of naive lymphocytes at HEVs, but unlike conventional lymphoid organs their secretion appears to be independent of LTα.36 IL-17–producing T cells33 and CCR7-dependent Tregs39 have been shown to be important for development of iBALT.
Knowledge of the role of iBALT in the lung is still evolving. BALT development appears to be part of the normal immune response to infectious antigens. The role of BALT expansion in a wide variety of chronic lung diseases such as hypersensitivity pneumonitis, chronic obstructive pulmonary disease, and idiopathic pulmonary fibrosis is less clear.40
 INTERSTITIAL LYMPHOCYTES
INTERSTITIAL LYMPHOCYTES
Lymphocytes are rarely seen in histologic sections of normal human lung, and there are no established techniques to study these cells as an exclusive, pure population. However, several investigators have prepared lymphocytes from human lungs extensively washed to remove airway surface cells using minced tissue and enzymatic digestion. The resulting population of pulmonary infiltrating lymphocytes in normal humans appears to be distinct from lymphocytes recovered from normal BAL.41,42 Specifically, approximately 20 × 106 mononuclear cells were found per gram of wet lung tissue; of these 70% were lymphocytes, of which 90% were CD2+ T lymphocytes. There was enrichment for memory T cells similar to that seen in LES, but the CD4/CD8 ratio among IL T cells was lower than that seen in blood or LES. Memory T lymphocytes from the interstitial compartment can be stimulated to produce cytokines, and to proliferate in response to IL-2 despite a decreased proliferative response to mitogens.43 Most, if not all NK activity in the lung has been localized to the interstitial compartment.44 The exact origin, fate, and function of interstitial lymphocytes are not known.
 INTRAVASCULAR LYMPHOCYTES
INTRAVASCULAR LYMPHOCYTES
The presence of this lymphocyte pool has been convincingly shown in animals, especially the pig.32 Experimental data includes lung perfusion studies showing the continued slow elution of lymphocytes from lung following elimination of red blood cells. The presence of an intravascular lymphocyte pool in humans has not been directly confirmed. However, labeled lymphocytes injected into humans are “held up” in the lung whether injected intravenously or intra-arterially (reviewed by Berman et al.6). These data confirm the more recently detected presence of homing receptors on the surface of lymphocytes that match cognate organ-specific chemokine expression.45 Therefore, this phenomenon likely represents margination of lymphocytes in capillaries due to adhesion molecule interactions. Complete phenotypic characterization of this pool in the human is unknown, as is the size and role of this pool in populating interstitial or epithelial lymphocytes.
 LYMPHOCYTE RECRUITMENT TO THE LUNG
LYMPHOCYTE RECRUITMENT TO THE LUNG
Lymphocytes are recruited to extravascular sites through a complex process involving adhesion to endothelial cells (ECs), release from adhesion, transendothelial migration, interaction with cellular matrix, and response to locally produced chemoattractants.6,46,47 This sequential process is characterized by an initial capture step that is mediated by selectins and integrins, followed by arrest/activation mediated by chemokine and cytokine receptors and other integrins.45,48 The role of chemokines has emerged as central to the regulation of tissue-specific lymphocyte homing and retention. These are low–molecular-weight proteins that share a cysteine repeat motif (e.g., C-C or C-X-C), near the N-terminus. As a class, these proteins have cationic charge, and therefore bind heparin.49 Chemokine function is mediated through cognate seven transmembrane-spanning receptors. There is significant promiscuity between chemokine receptors and their ligands, such that any specific chemokine may have several ligands and vice versa. Because of their physicochemical properties, the chemokines may bind to heparin-like regions of EC membrane receptors, preventing dilution of locally produced chemoattractant signal by blood flow and facilitating concentration gradients, a phenomenon known as haptotaxis. Chemokines have been shown to enhance both adhesion to ECs and endothelial transmigration of multiple leukocyte types including T-cell subsets and monocytes. Recently, chemokines have also been found to act as accessory growth factors for T-lymphocytes.50 While CXCL15 (lungkine) appears to be selectively expressed in mouse lung, there does not appear to be a unique human lung–specific lymphocyte directed chemokine profile. However, certain chemokines (see below) are preferentially expressed in TH2 and TH1 type immune responses in the lung.
The interaction of adhesion molecules expressed on blood T cells with complementary adhesion molecules on EC is the critical first step to T-cell emigration from the blood. This step is closely regulated at the level of expression of adhesion molecules by T cells at different stages of development, and by transiently increased adhesion molecule function following T-cell activation. Similarly, expression of adhesion molecules by EC may be increased markedly by organ site and location, or by proinflammatory cytokines, especially tumor necrosis factor (TNF)-α, IL-1, and interferon-γ (IFN).51–54 Treatment of EC with these cytokines markedly alters the adhesiveness of EC for leukocytes, including certain subsets of T cells. The result of such EC activation by local production of cytokines or other factors is that the exit of T cells from the blood is not random, but rather is restricted to sites such as lymphoid tissues, mucosal sites, or tissue sites of inflammation.
The specific events involved in T-cell transendothelial migration have been dissected at the cellular level, while their characterization in human lung is incomplete. T lymphocytes first appear to “roll” along the endothelium, an interaction which requires loose adhesion via T-cell α4β1 integrin/endothelial VCAM-1 and P-selectin/PSGL-1.15 A signal is required to produce formal “capture” via other integrin molecules, particularly lymphocyte LFA-1 (αLβ2 integrin) interaction with ICAM-1 or other ligands on endothelium. This latter signal enhances the avidity of integrins for ligand, strengthening adhesion. Other interactions, including decay of integrin affinity, homotypic interaction of T cell and endothelial PECAM (CD31) at EC junctions, and release of matrix degrading enzymes, permit release of firm adhesion and T-cell migration into matrix.47 Preliminary evidence in TH2-cell migration suggests that this step is mediated by T-cell CCR3/endothelial CCL11 and CCR4/CCL17, respectively.16,55,56
T cells are constantly recirculating from blood to tissue and back, with an average half-life in the blood of only about 18 hours.57 In addition, the sites of migration in vivo appear to be quite different for naive/virgin cells, as opposed to previously activated memory cells. Due to a high level of CCR7 expression, naive cells preferentially traffic to lymphoid tissue where they are likely to encounter antigen,58 while memory cells traffic to nonlymphoid tissues such as skin or lung.53,59
The normal lung vasculature may have unique properties that facilitate the retention of circulating lymphocytes. Whether injected intravenously or intra-arterially, labeled lymphocytes are held up in the lung disproportionately in comparison to other organs. Thus, this is not just a “first-pass” clearance effect from capillary passage.6 Several investigators have found that antibodies to adhesion molecules, particularly lymphocyte function–associated antigen (LFA)-1, decrease lymphocyte retention in the lung. However, capillary size may determine trapping of activated cells since cytoskeletal changes occurring coincident with activation reduce cellular deformability; this has been shown to be a significant force in the lung trapping of activated neutrophils and monocytes. Lymphocytes also become larger, and lose deformability with activation, but the role of this process in lung retention of lymphocytes is not yet defined.
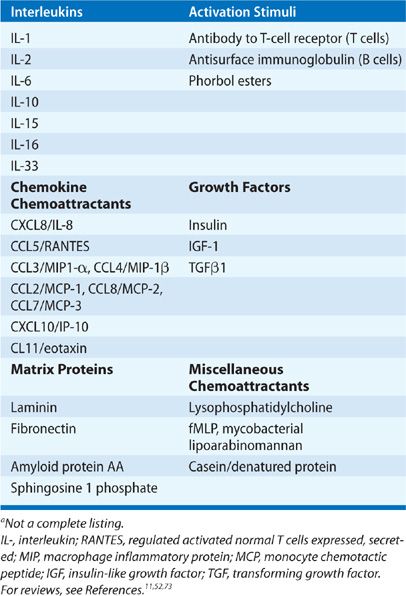
Lymphocyte chemoattractants represent another step in the regulation of the exit of adherent or trapped T cells from the capillary circulation. As noted earlier, varied T-cell chemoattractants have been described which are relevant to the lung, many of which also alter the growth and activation of T cells (reviewed in References6,49,50,60). A partial listing of known chemoattractants may be found in Table 21-3. In lung diseases multiple T-cell chemoattractants have been found in BAL or in tissue specimens, including the chemotactic growth factors IL-2,29,61 IL-16,62 IL-33,63,64 insulin-like growth factor I13, the C-X-C chemokine IL-8 (CXCL8), and the C-C chemokines macrophage chemotactic protein (MCP)-1 (CCL2), RANTES (CCL5), and macrophage inflammatory protein (MIP)-1α (CCL3).62,65,66 These chemokines have multiple cellular origins, including macrophages, ECs, and epithelial cells. IL-2 is presumed to be of lymphocyte origin while IGF-1 and IL-16 have multiple potential cellular sources including T cells, eosinophils, and epithelial cells.67,68 IL-33 is an epithelial-derived alarmin which in addition to being a chemoattractant to TH2 cells via ST2/IL1RAcP receptor also attracts ILC2, eosinophils, and mast cells. Its central role in TH2 immune deviation in asthma in response to epithelial cell necrosis to a variety of injuries is becoming increasingly appreciated.69–72 It has also been noted that CXCL9, 10, and 11 appear to be important in TH1 responses in the lung, but their expression may relate more to induction by interferon-γ rather than a lung-related phenomena.
TABLE 21-3 Lymphocyte Chemoattractantsa
Recruitment of lymphocytes to the lung along chemotactic gradients is a complex phenomena involving multiple chemotactic factors, gradients, and responding cell receptors. The final complexion of the lymphocyte population in any compartment is therefore the sum of the responsiveness of each phenotype sequentially responding to and then releasing from chemotactic gradients to respond again. The hierarchy of responses involves regulation of expression of receptors and responsiveness, a phenomena termed chemotactic factor receptor cross desensitization. This can explain retention of T cells (lack of responsive chemotactic receptors) and relocation (sequential responsiveness to induced or constitutive chemotactic gradients). Examples of these modulatory steps are present in human74–76 and mouse T cells of all phenotypes and likely this is one of the prime factors for accumulation of T cells in organized lymphoid tissues in the thorax and lung (e.g., lymphadenopathy) during infection and inflammation. Moreover, chemotactic gradients and receptor desensitization can be induced by exogenous chemotactic factors of infectious origin, which can in turn regulate responses to endogenous chemotactic factors. For example, HIV-1 gp120 can downregulate the responsiveness of thoracic lymph node lymphocytes to sphingosine-1-phosphate, resulting in lack of normal egress.74,75,77
 LYMPHOCYTE FUNCTION IN THE LUNG
LYMPHOCYTE FUNCTION IN THE LUNG
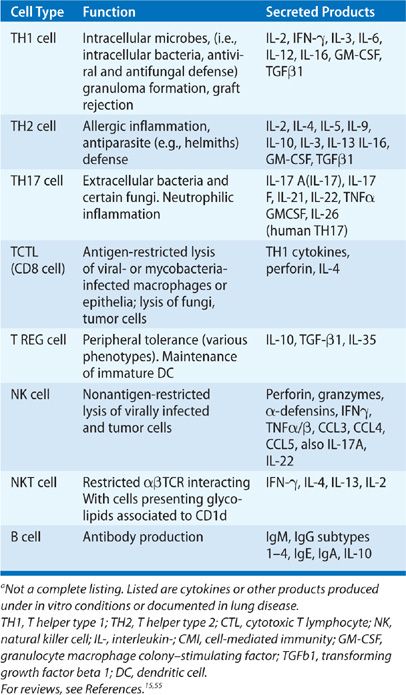
Lymphocytes in the lung serve four major functions: (1) antibody production; (2) cytotoxic activity including lysis of virally infected cells, cells that have bound antibody, and tumor cells; (3) cytokine production; (4) immune tolerance. These functions are summarized in Table 21-4 and in a number of references cited.5,49,78
Antibody production in the lung by B lymphocytes serves to bind antigen and facilitate inactivation of bioactive material and phagocytosis by macrophages. Mucosal IgA is of particular interest in its active transepithelial transport to the bronchial lumen. Antibody production by lung B cells has been extensively studied in mouse lung. Following challenge, antigen is removed to regional lymph nodes by motile phagocytic cells (macrophages and DCs) where optimal activation of T and B cells occurs. Activated cells relocate into the circulation and migrate into the lung at areas of inflammation, and local antibody production results. Rechallenge with antigen results in a more rapid local response derived from resident memory cells.5,6,79 While the duration of pulmonary memory lymphocytes is unknown, systemic B lymphocytes can persist for over 100 days in the absence of antigen,80 as is the case with memory CD4+ and CD8+ T cells.81
The lung contains multiple types of cytotoxic cells, including NK cells (not antigen restricted), antigen-restricted cytotoxic cells, and cells exhibiting antibody-dependent cytotoxicity. One unusual aspect of lung cytotoxic cells is the pre-eminence of CD3+ cytotoxic T cells with NK activity (nonantigen-receptor–mediated killing of tumor cell targets). This is in contradistinction to the blood, where the majority of cells expressing NK activity are CD3–.5 These natural killer T cells (NKT cells) make up less than 0.2% of human peripheral blood T cells and, contrary to an earlier report,82 are low in number in bronchial tissue and BAL in health and disease. In asthmatics, it is approximated they account for, at most, 1.7% and 0.2% in bronchial tissue and BAL, respectively.83 They have an invariant TCRα–receptor chain receptor (variable [V] and joining chain [J] Vα14 Jα18 in mice and Vα24 Jα18 in humans) combined with a limited, but not invariant TCRβ-chain repertoire (Vβ8.2, Vβ7, or Vβ2 in mice and Vβ11 in humans), and conventionally are referred to as iNKT or type 1 NKT cells. They recognize foreign and self-glycolipid antigens presented by MHC-class-I-like CD1d antigen-presenting molecule (reviewed by Godfrey, Stankovic, and Baxter84). While a role for iNKT cells in host defense to a variety of infectious antigens (e.g., Cryptococcus neoformans)85 and in modulating the inflammatory response in asthma86 has been suggested, details of their function have to be elucidated.
TABLE 21-4 Function of Lung Lymphocyte Subpopulationsa
NK activity is found in the interstitial compartment of lung T cells and NK cells are present among LES accounting for approximately 10% of tissue lymphocytes. The majority (80%) are phenotypically the cytotoxic subset (CD56dimCD16high) in humans and resting pulmonary NK cells have been found to be functionally impotent.44,87–89 The impairment of cytotoxic capacity of resting pulmonary NK cells in BAL and lung tissue is believed to be due to the suppressive effects of cytokines such as TGFβ and other mediators such as prostaglandins from AMs, pulmonary surfactant90 and respiratory epithelial products such as IL-15.91 The importance of this regulation of NK cells in the lung is exemplified in certain genetic deficiencies which result in either chronic activation of NK cells leading to granulomatous inflammation or recurrent viral and bacterial infections that can involve the upper and lower respiratory tract.92–94
Cytokine production by lung T helper cells (TH) has emerged as a major focus of investigation in lung inflammation. In contrast to antibody production by B cells, T cells produce cytokines. Broad ranges of cytokines have been documented to be produced by lung T cells in inflammatory disease (Table 21-4). In general, the complexion of the inflammatory response correlates with the cytokines produced by T cells, suggesting that T cells orchestrate many inflammatory responses.
Activated T-helper (TH) cells produce a distinct spectrum of cytokines.95–97 The repertoire of a single T cell to produce cytokines appears to be limited and stereotyped, depending on the circumstances of activation. According to data accumulated in mice, naive T cells produce mainly IL-2 in response to activation. After proliferation and switch to memory cell phenotype, T cells differentiate and produce one of four major clusters of effector cytokines, the best characterized being TH1 (interferon-γ, IL-2), TH2 (IL-4, IL-5, IL-10, IL-13), TH17 (IL-17, 23, 25), and Treg (TGFβ, IL-10, PGE). These TH phenotypes share mutually exclusive transcriptional programs, regulated by the transcription factors T-bet (TH1) and GATA-3 (TH2), RORγ/STAT3 (TH17), and FoxP3 (Treg), respectively.98–100
Historically, the division between TH1 and TH2 phenotypes was derived by exclusion of the alternate type and led to confusion in the distinction between these phenotypes particularly in humans where the distinction is not as predictable as in the mouse.95 These two major phenotypes roughly conform to polarized expressions of cell-mediated immune responses: granuloma formation with activation of mononuclear phagocytes and production of opsonizing IgG2 antibody (TH1); or optimal antibody response including IgE formation, often with associated eosinophilia (TH2).
Certain immune responses in the lung are dominated by either a TH1 or TH2 response, while others are mixed. For example, in human asthma T cells producing TH2 cytokines predominate,101 but IFN-γ producing cells are found in the airways suggesting a mixed response.102 In contrast, granulomas at sites of tuberculin reactions in skin show evidence for production of IFN-γ and IL-2 but not IL-4.103 In leprosy or leishmaniasis an ineffective host reaction is associated with a TH2 response, and an effective granulomatous response is associated with a TH1 response. In addition, treatment of ineffective responses to leishmania with IFN-γ have been reported to increase the efficacy of chemotherapy.96 In sarcoidosis, airway and granuloma cells, particularly activated CD4+ (HLA-DR+) T cells, have been found to produce both IL-2 and IFN-γ, suggesting the predominance of a TH1 response. There is considerable cross-regulation of TH1 and TH2 subsets even after commitment to production of these cytokines, leading to the general concept that the character of an immune response as well as its termination may depend on the sequential predominance of TH1 or TH2 responses. Indeed, the functional distinction between TH1 and TH2 cells is less distinct in humans as it is in the mouse. As a result of these earlier studies, it is now clear that other T-cell phenotypes participate in all these inflammatory events in the lung beyond traditional TH1 and TH2 dichotomy.
First, there is an important role of T cells (and T-regulatory cells in particular) in mediating mucosal tolerance and immune homeostasis.104,105 This function in the mucosa is directly related to their interaction with mucosal DCs.78,106,107 While the phenotypic classification of these cells is evolving, the peripheral pool of regulatory T cells (Tregs) is a mixture of natural thymus–derived Tregs (nTregs) and induced Tregs (iTregs), which originate from naive CD4+ T cells under conditions of low antigenic stimulation or by cytokines primarily transforming growth factor beta (TGFβ). While the relative contribution of each type of Treg to immune suppression and homeostasis is not clear, the fact that their TCR repertoire is different suggests a nonredundant complementary role.108 There appear to be multiple phenotypes within the Treg population with overlapping mechanisms of action. However, the transcriptional factor forkhead box protein 3(Foxp3) appears essential for the suppressive function of this T-cell subset. Deletion of Foxp3 in mice (Scurfy) and humans (IPEX syndrome) results in a lethal condition of autoimmunity and inflammation emphasizing the critical role of Tregs in immune homeostasis and IL-2 in the growth and expansion of Treg. The majority of studies examining the regulatory potential of T cells in inflammatory lung disease, however, have been in ex vivo manipulation, depletion, and adoptive transfer experiments in the mouse.109,110 The evidence for intraparenchymal (i.e., mucosal or interstitial) human Treg cells is not direct, but the requirement for maintenance of peripheral tolerance in the lung and systemic inducibility of tolerance to inhaled antigen111 provides the basis for their speculated presence in the epithelium. In addition, intratracheal treatment of mice with the immunomodulatory cytokine IL-16112,113 elicits an expansion of lung CD4+CD25+ T cells.114
TGFβ is required for the development of iTregs, but dual stimulation of TGFβ with either IL-4 or IL-6 (IL1β in humans) can alter the cytokine profile of peripheral T cells from suppressive Tregs to proinflammatory TH9 or TH17 cells, respectively. Similar to TH1 and TH2 development, TH9/TH17 and Treg cell development is cross-regulated at the transcription factor level, an important mechanism for maintaining immune homeostasis in the periphery.115 At the functional level, this requirement of TGF-β for the induction of both Foxp3+ Tregs and TH17 cells provides a system for efficient balance between tolerance and immunity. In the steady state, TGFβ induces Foxp3 and Tregs, inhibits inflammation, and maintains self-tolerance, but once IL-6 is produced by innate immune cells in response to microbial triggers, Treg generation is prevented and the function of nTregs is suppressed while TH17 cells are induced to produce a strong proinflammatory response characterized by neutrophilia.115
While a distinct functional role of predominantly IL-9 secreting TH9 cells in the lung is unknown, the TH17 cell cytokine cluster (IL-17A, IL-17 F, IL-22, and IL-21) is understood to help protect the lung against extracellular bacteria and fungi.116 However, the observed elevations of IL-17A and IL-17F in sputum and blood/BAL in asthma117,118 indicate that TH17 cells contribute to chronic inflammatory disease as well. TH17 cells, like TH1 and TH2 cells, accomplish some of their role by recruiting cells of the granulocytic–monocytic lineage: TH1 cells recruit monocytes/macrophage, TH2 cells recruit eosinophils, basophils, and mast cells, and importantly TH17 cells recruit neutrophils. Given that up to 50% of asthmatics are nonatopic116 and IL-17 mediates neutrophilic inflammation, there has been interest in understanding TH17 role in difficult to treat asthma. Interestingly, in vitro experiments have shown that the neutrophilic airway inflammation and bronchial hyperresponsiveness caused by TH17 cells was steroid resistant, consistent with the clinical phenotype.119 While sarcoidosis is classically thought of as a TH1 disease, IL-17–producing cells are also elevated in peripheral blood and bronchoalveolar lung fluid of these patients.120 Moreover, both single IL-17A+ and double IL-17/IFNγ+ cells have been reported in sarcoid granulomatous tissue.121 Thus TH17 cytokine cluster helps understand the heterogeneity in classical TH1 and TH2 disease in the lung.
However, T cells are not the only source of IL-17, innate lymphoid cells (ILCs) such as NKT cells, NK cells and γδ T cells produce IL-17.115 There is great interest in understanding the contribution of ILCs to immune homeostasis and pathology in the lung. Three features define ILCs: the absence of recombination-activating gene (RAG)-dependent rearranged antigen receptors; a lack of myeloid cell and DC phenotypic markers; and their lymphoid morphology. While the prototypical ILC population is NK cells (see previous section), several other distinct ILC populations have recently been described, all of which appear to originate from a common lymphoid (Id2+) progenitor cell and all, like conventional T cells, produce cytokines.
A particular source of confusion is the number of different names that have been used to characterize these different ILC populations. For example, ILCs that produce TH2-cell–associated cytokines have been variously called natural helper cells,122 nuocytes,123 and innate helper 2 (Ih2) cells124 without clear evidence of uniqueness. A functional nomenclature system based on cytokines produced125 has recently been proposed: Group 1 ILCs produce IFNγ, Group 2 produce type 2 cytokines (including IL-5 and IL-13), and Group 3 ILCs produce IL-17 and IL-22. ILCs are changing our view of how lymphocytes function in lung inflammation. For example, ILC2 have been shown to contribute IL-13 not IL-4 production in ovalbumin-induced lung inflammation mouse models126,127 and in IL-13 –/– mice, which are resistant to allergic lung inflammation and virus-induced airway hyperresponsiveness transfer of IL-13 expressing ILC2 s was sufficient to restore airway hyperresponsiveness in both models126,128 A human equivalent of ILCs have been found in patients with chronic rhinosinusitis.129
B-lymphocyte biology has also significantly advanced in recent years and now B cells are known to be comprised of different populations and proven to be pleiotropic in function (Ref.130 summarizes the current understanding of B-cell lineages in the lung.) Briefly, in mice the majority of B cells are B2 B cells, which originate from the adult bone marrow precursor, traffic through peripheral lymphoid tissue, interact with cognate antigen, and after activation develop into either memory B cells or antibody-secreting plasma cells. In the human lung, the majority of B cells are these mature naive B2 B cells and they reside in BALT (see previous section) or draining lymph nodes. In mice, there is a family of innate-like B cells: marginal zone B cells, B1α and B1β B cells that contribute to rapid immune responses against pathogens and other stimuli. B1 B cells are a small self-renewing subset of B cells that originate from the fetal liver and secrete most, if not all, natural antibodies (IgM and IgA isotype) in the apparent absence of antigenic challenge. Natural antibodies are often polyreactive and bind to foreign antigens as well as to self-components (ex rheumatoid factor) and are important for early pathogen recognition and maintenance of tissue homeostasis.131 B1 B cells constitute a minor fraction of the spleen and secondary lymphoid tissues but are enriched in the pleural and peritoneal cavities. It is not known whether B1 B cells residing in the pleural cavity migrate into the lung. Human B1 B cells do exist and are found to have marker profile of CD20+CD27+CD43+CD70– and could either be CD5+ or CD5–.132
The central role of B cells is the production of immunoglobulins, both within the parenchyma and for export to the mucosal surface of the airway, in the case of polymeric IgA and IgM. In addition, B cells are now known to function as antigen-presenting cells and producers of both inflammatory and regulatory cytokines (i.e., IL-10–producing B-regulatory cells [B-regs]). Very little is known about either of these functions in the lung by B cells. Much of disease immunopathology in the lung is based on T-cell biology, but there is an expanding appreciation of B-cell contribution to diseases such as hypersensitivity pneumonitis, COPD, autoimmune connective tissue diseases, and idiopathic pulmonary fibrosis (see the review by Kato et al.130 for details). But overall, all these diseases illustrate the potential for the lung to generate an exuberant B-cell response to antigen, either inhaled or self-antigen.
 LYMPHOCYTE ACTIVATION IN THE LUNG
LYMPHOCYTE ACTIVATION IN THE LUNG
T lymphocytes are designed to require specific (antigenic) signals for activation, restricting their involvement in inflammation to situations where antigen overwhelms the mucociliary escalator and macrophage and neutrophil defenses. Lymphocytes are activated following engagement of an antigen receptor of remarkably fine specificity. This receptor is unique to a given lymphocyte clone, and is generated by recombination of gene segments in the antibody (for B cells) or T-cell–receptor genes. B-cell receptors consist of single membrane–spanning antibody molecules of the same specificity as the B cell, while T-cell receptors consist of a heterodimeric receptor (dimers of alpha and beta or gamma and delta chains). The T-cell receptor is highly antigen specific and has structural homology to the immunoglobulin molecule. Lymphocytes are activated by cross-linking of membrane antibody by antigen (B cells) or by engagement of the antigen receptor by antigen bound to major histocompatibility complex (MHC) molecules on the surface of so-called antigen presenting cells (APCs), also known as accessory cells. APCs provide many “accessories” for T-cell activation including: (1) a source of MHC molecules to which antigen can bind; (2) internalization and “processing” of antigen including protease digestion into antigenic fragments; (3) multiple cell adhesion molecules which bind to complementary adhesion molecules on T cells and serve to strengthen T-cell–accessory cell interactions and transduce activation signals required for optimal lymphocyte activation; and (4) production of cytokines that amplify activation including IL-1 (reviewed by Cruikshank et al.112).
APCs in the lung include pulmonary macrophages of all varieties, however, DCs and Langerhans cells are most efficient in this function (see below). These cells express important “accessory” cell adhesion molecules important to accessory cell function, including ICAM-1, LFA-2, LFA-3, and the CD28 ligands B7–1, B7–2, and CTLA-4.68,133,134 Other cells, which may be induced to express Class II MHC molecules, may also act as weak accessory cells. Such cells include local B cells, epithelial cells, smooth muscle cells, and fibroblasts. Uncommitted naive T cells require intense accessory cell interaction to be activated by antigen, while previously activated (memory) T cells require less accessory cell input and might be influenced by interaction with such weak accessory cells. Due to naive CCR7+ T cells’ inability to cross postcapillary venules, as mentioned earlier most lung T cells are memory cells, which suggests that the relatively weak lung accessory cells may indeed play a role in T-cell activation in the lung inflammatory response (Table 21-5).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


