Leukocyte Accumulation in Pulmonary Disease
INTRODUCTION
Mediators produced during inflammatory/immune responses dictate the severity and intensity of pulmonary disease. The profile of inflammatory leukocyte populations accumulating in inflamed tissues is initiated by cytokine-induced expression of adhesion molecules on the vascular endothelium. Endothelial adhesion molecules include intracellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), as well as E- and P-selectins that initiate, and in some cases, mediate the migration of leukocytes into tissues. Subsequently, leukocyte adherence to the endothelium is followed by leukocyte migration into the inflamed tissue, directed by chemotactic molecules at the site of the inflammatory/immune response. Upregulation of these early response mediators is crucial for the initiation of early events that regulate the inciting agent, whether it is infectious or noninfectious in nature. However, the continuous over-production of these mediators can lead to destructive, pathologic consequences due to the continued recruitment and activation of disease-specific leukocyte populations. In human lung, inflammation-induced damage can be observed in numerous inflammatory diseases, including both acute and chronic disease settings. In this chapter, we will examine the mediators that promote inflammatory diseases in lung and outline how specific leukocyte populations can contribute to pulmonary pathology.
LEUKOCYTE ADHESION AND MIGRATION INTO THE LUNG
Important considerations in the biology of leukocyte adhesion and migration into the lung are discussed below.
 SELECTIN AND ADHESION MOLECULES IN LUNG INFLAMMATION
SELECTIN AND ADHESION MOLECULES IN LUNG INFLAMMATION
The release of early response mediators leads to the upregulation of selectins (E and P) and other adhesion molecules (ICAM-1, VCAM-1, etc.) on surfaces of vascular endothelial cells within the site of inflammation.1–7 Initially, selectin molecules (P and E) are quickly upregulated on the vascular endothelium and initiate “rolling” of leukocytes on activated endothelium through Ca2+-dependent recognition of cell surface carbohydrates of the sialyl Lewis X family and related oligosaccharides. Initial and rapid expression of selectin molecules results in a slowing of leukocyte velocities in the circulatory flow, allowing additional interactions to proceed.8–11 However, such interactions are required to ultimately allow firm adhesion of leukocytes to endothelial cells. Once the leukocytes have begun the selectin-mediated rolling process, they must next go through a series of activation events to allow them to firmly adhere to other adhesion molecules. Leukocytes ultimately bind firmly to the vascular endothelium via β-integrin receptors on leukocyte surfaces, resulting in a very rapid increase in binding affinity, and engagement of other molecules that are upregulated during inflammatory responses on the vascular endothelium. A number of β-integrin adhesion molecules play a role in the migration process, and they are differentially expressed on subsets of leukocytes.12–14 The β1α4 integrins (VLA-4), expressed primarily on mononuclear cells and eosinophils, have been shown to bind to vascular cell adhesion molecule-1 (VCAM-1), while β2-integrins (CD11/CD18) are expressed on all leukocytes and bind varyingly to intracellular adhesion molecules-1,2,3 (ICAM-1,2,3), the first of which is highly expressed on endothelial cells. These families of adhesion molecules are able to facilitate leukocyte binding to the activated endothelium and can further dictate the type of leukocytes that bind and extravasate into the inflamed tissue. For example, while neutrophils rely on CD11/CD18 binding to ICAM-1, eosinophils depend upon VLA-4/VCAM-1 interactions to firmly adhere to the endothelial cell surface. Once firmly adherent, leukocytes then enter the tissue following chemotactic gradients through a series of detachment/readherence events typified by the polar expression of integrins specific for the adhesion molecules contained on surfaces of mesenchymal-derived cells.
Cell-to-cell communication during inflammatory events is mediated by cytokines that initiate, maintain, and regulate the inflammatory responses, dictating the intensity of the inflammatory response. The early response cytokines, IL-1 and TNF appear to play a pivotal role in the induction of inflammatory responses through the initiation of cytokine cascades.15–19 The exuberant production of IL-1 and TNF may lead to multisystem injury and systemic complications, as exemplified in septic shock syndromes. As indicated earlier, IL-1 and TNF initially upregulate selectin (E-selectin) and other adhesion molecules (ICAM-1, VCAM-1) needed for the first step of leukocyte extravasation into tissues. In addition, IL-1 and TNF upregulate other inflammatory cytokines (e.g., IL-6) involved in the chemotactic responses of leukocytes into inflamed tissues. Interestingly, the type of cytokine expressed can dictate the nature of the inflammatory response based upon the adhesion molecule that it induces. For example, while TNF and IL-1 are critical for upregulation of ICAM-1 that facilitates neutrophil and monocyte adhesion, IL-4 produced during allergic responses preferentially upregulates VCAM-1. This adhesion molecule mediates eosinophil adhesion. Thus, the inflammatory/immune cytokine environment in lung can tailor the initiation and adhesion interactions for a particular leukocyte recruitment profile. The production of one of a number of classes of chemotactic factors is required for the movement of leukocytes from the vascular compartment to the extravascular compartment of the lung. We will next describe and characterize the function of chemotactic mediators that are expressed in the lung during specific disease conditions.
 CHEMOKINES AND GPCR SIGNALING IN LEUKOCYTE ADHESION
CHEMOKINES AND GPCR SIGNALING IN LEUKOCYTE ADHESION
The rapid change in affinity of the β-integrins rely on two events: (1) Initial binding to selectin molecules that activates Syk and MAPK, allowing β-integrin to reach an intermediate affinity state; and (2) activation of leukocyte expressed G-protein coupled receptors (GPCR) on the surface of leukocytes.20–27 The GPCR ligands, usually chemokines bound to the endothelium via glycosaminoglycans (GAGs), initiate rapid Ca2+-dependent β-integrin activation.28,29 The β-integrin activation results in a conformational change in its extracellular domain, allowing the active binding site for the putative adhesion molecule to be accessible. If the adhesion molecule is also upregulated and expressed on the activated vascular endothelium, the leukocyte rapidly and firmly adheres and spreads along the endothelial surface due to additional actin polymerization, which is also induced during chemokine-GPCR activation events. Thus, the leukocyte very rapidly transitions from a rolling leukocyte to a cell that is firmly adherent, resulting subsequently in transmigration into the tissue. In addition to the GPCR signal, it also appears that the shear stress experienced by leukocytes also plays a role in development in firm adhesion events. These interactions are described in Figure 23-1.
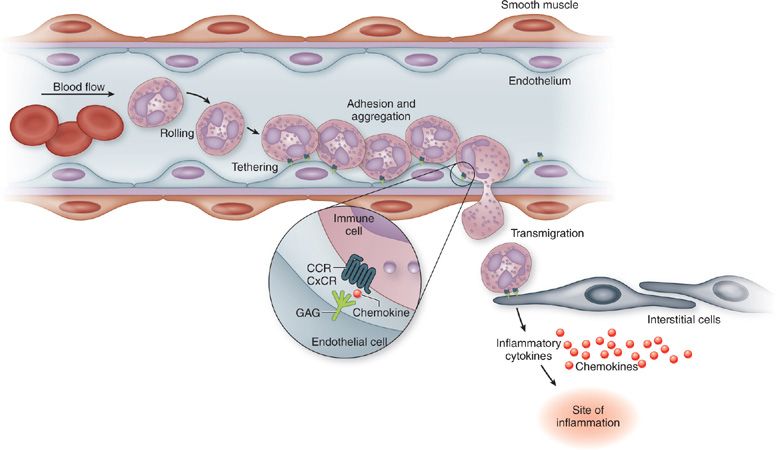
Figure 23-1 Leukocyte migration requires coordinated interactions between adhesion molecules and chemotactic molecules.
A significant amount of research has occurred in recent years, defining critical signaling events that occur during the early stages of leukocyte migration, including selectin-mediated rolling, GPCR-mediated integrin binding, actin polymerization, and leukocyte polarization. One of the most critical events in the transition of leukocytes from rolling to firm adhesion and extravasation is engagement of endothelial bound chemokine ligands with appropriate GPCRs. Chemokine binding activates the Gi-coupled protein-mediated phospholipase C (PLC), which results in inositol-1,4,5 triphosphate (IP3) formation as well as diacylglycerol (DAG), triggering intracellular Ca2+ increases due to translocation from the ER.30–32 This signaling cascade initiates the activation of Rho GTPases, Rap-1 and Talin1, subsequently mediating integrin affinity increase for their adhesion molecule ligands and allowing clustering following binding. Blockade of events at any stage along this activation pathway can inhibit the β-integrin–mediated adhesion, and interrupting firm adhesion to the activated endothelium.
 CHEMOATTRACTANTS AND MIGRATION INTO THE LUNG
CHEMOATTRACTANTS AND MIGRATION INTO THE LUNG
Below is discussed the role of various chemoattractants in the immune response against pathogens, including complement, arachidonic acid and its derivatives, and chemokines.
Complement
The complement activation cascade plays a significant role in the innate immune response against pathogens, but continual or excessive activation during inflammatory or infectious responses can lead to severe tissue injury. The initiation of the complement cascade can be accomplished via multiple mechanisms, including antibody–antigen complexes, bacterial products, toxins, and lectins (Fig. 23-2). Of the complement activation products, fragments from C3 and C5 have the most profound effects on the inflammatory response. The split products of C3, C3a and C3b, are generated by C3 convertase (other products of C3 cleavage [iC3b, C3d, C3g]) have significant activating roles in the inflammatory pathway. C3a is an anaphylatoxin that induces the activation of mast cells/basophils resulting in mediator release, all of which appear to have direct and indirect effects on vascular permeability.33–35 C3b acts as a potent opsonizing component, binding to bacteria and allowing accelerated phagocytosis and clearance of pathogens via the C3b receptor on neutrophils and macrophages (Mac-1 [CD11b/CD18]). The split products of C5, C5a and C5b, can subsequently be induced through the sequential participation of C3b and C5 convertase. Similar to C3a, but much more potent, C5a is an anaphylatoxin that interacts with its two receptors (C5aR, C5L2), causing mast cell and basophil degranulation and activation of neutrophils, which collectively induces immediate changes in vascular permeability.34,36–40 In addition, C5a stimulates vascular smooth muscle contraction and has neutrophil chemotactic and activating characteristics that promote directed migration of these leukocytes toward a concentration gradient.41,42 C5a can also stimulate neutrophil oxidative metabolism, granule discharge, adhesiveness to vascular endothelium, and assembly on the neutrophil surface of NADPH oxidase (NOX2). C5a can directly stimulate endothelial cells in a G-protein receptor-dependent fashion to cause signal transduction events resulting in increased intracellular Ca2+, induction of superoxide (O2–), and expression of P-selectin.43–45 C3a lacks these activities. Altogether, the functions of C3 and C5 split products indicate that they are potent inflammatory mediators.
Elevated complement component levels in plasma have been described with several pulmonary diseases, including sarcoidosis, idiopathic pulmonary fibrosis (IPF), acute respiratory distress syndrome (ARDS), and chronic obstructive pulmonary disease (COPD).46,47 Not only can complement initiate GPCR-mediated leukocyte migration but it appears complement activation products can direct the development of immune responses. This can be accomplished via several pathways during initiation of immune responses by regulating IL-12 production.48 Specifically, C3a appears to drive IL-12 production that then favors type 1 immune responses,49 while C5a downregulates IL-12 and allows IL-4–mediated type 2 immune responses to be induced.50 Interestingly, recent studies in C3–/– mice have shown that Th17 responses are also regulated by complement.51 In contrast, another study suggested that, when C5aR was blocked, there was an exacerbated allergic response that also was dependent upon increased IL-17,52,53 suggesting that the previous study with C3–/– mice was due to inhibition of downstream C5a activation. Thus, while early complement-induced response is critical for containment of infectious organisms, it may be especially important during sensitization to various antigens in determining the phenotype of the pulmonary immune response that will govern the severity of disease outcomes.
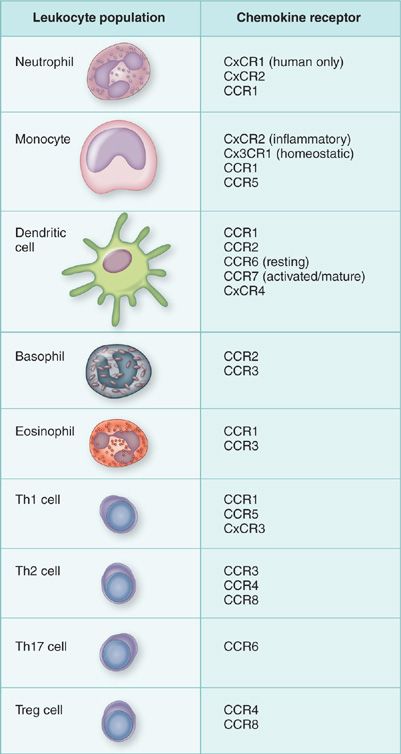
Figure 23-2 Preferential expression of chemokine receptors on leukocyte subsets can lead to preferential recruitment during pulmonary disease.
The receptors for C3a and C5a have been a source of intense research over the past several years and have led to the resurgence of interest in potentially blocking specific responses during pulmonary disease. The distribution of these receptors depicts their broad role in innate and acquired immune responses. Both C3aR and C5aR are present on alveolar macrophages, DCs, and mast cells, and are also present on sentinel cell populations in the lung that provide important cues for the immediate and prolonged determination of effective immune responses.54 In particular, activation of these cell populations have been assessed for the differential activation consequences of C3a and C5a. At the same time the expression of C5aR on neutrophils allows an immediate and efficient migration into the lung and activation at the site of inflammation. C5aR is also displayed on cells involved in chronic immune responses, such eosinophils recruited during allergic responses. Thus, activation of the complement system can have potent effects on both acute inflammatory responses as well as chronic inflammation, both of which can lead to long-term pulmonary dysfunction. Interestingly, a second C5a receptor, C5L2, binds C5a and C5a des arg extremely efficiently, but C5L2 has no linkage to G proteins.55 Research examining the possible implications of this second receptor for modulating C5a-mediated responses by competitively binding up C5a during inflammatory responses is the subject of focus from several laboratories. While the function of this second C5a receptor has been controversial, C5L2 appears to mediate several pathologic events, including events in sepsis and perhaps diabetes.56–60
Role of Arachidonic Acid and its Products in Lung Responses
The production of arachidonic acid (AA) followed by its enzymatic processing produces a number of lipid mediators that have long been known to be associated with acute and chronic inflammatory responses in the lung.61–63 Phospholipase A2 degradation of AA leads to its breakdown into platelet activation factor (PAF) that can be immediately processed by 5-LO into 5-HPETE and further processed by 5-LO into leukotriene A4 (Fig. 23-3). 5-HPETE can alternatively be processed into 5-HETE by peroxidases. Leukotriene A4 can be further processed into LTB4 by LTA4 hydrolase or LTC4 by LTC4 hydrolase, followed by continued processing of LTC4 into LTD4, and finally, LTE4. These latter metabolites, C4, D4, and E4 are known as the cysteinyl leukotrienes that are best known to cause airway contractility, causing decreased lung function during large airways disease, such as asthma. Targeting the cysteinyl leukotrienes for blockade has provided significant relief to particular subsets of asthmatic patients.64
Of the AA metabolites, PAF and LTB4 have been characterized for their chemotactic activity, promoting migration of leukocytes into sites of inflammation. These mediators were originally identified as potent neutrophil chemoattractants and were associated with acute inflammation-induced airway damage. PAF has a broad range of specificity in its ability to induce leukocyte chemotaxis, since it induces the recruitment of not only neutrophils, but also of monocytes, lymphocytes, and eosinophils.65,66 During allergic diseases, PAF may have a role in augmentation of eosinophil responses.67 The actions of PAF on endothelial cells indicate that PAF has a direct role in upregulation of selectin and adhesion molecules and can induce the release of superoxide anion (O2–). Instillation of PAF into human, monkey, or guinea pig airways induced an immediate LTC4-independent bronchoconstrictor response, suggesting a possible role in development of pathophysiology in asthmatic responses. Overall, these studies indicate that the signals provided by PAF are not only chemotactic but can also participate in the augmentation of immune/inflammatory pathways in lung. However, over the years several PAF specific inhibitors have been developed, but they have failed to demonstrate effective blocking or attenuating inflammatory responses in human lung.
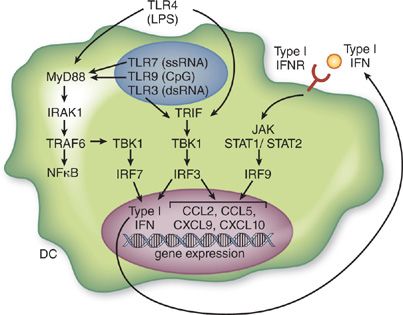
Figure 23-3 Inflammatory gene activation by TLR and type I IFN leads to enhanced leukocyte recruitment.
While leukotrienes in general have broad effects on inflammatory responses, LTB4 is one of the most potent neutrophil chemotactic molecules known and induces O2– production. LTB4 can also act to recruit other leukocyte populations, such as monocytes and eosinophils.68,69 In eosinophil chemotactic assays, LTB4 is more potent than PAF in the activation and degranulation of eosinophils. LTB4 has been found in many disease states, including psoriasis, bacterial peritonitis, inflammatory bowel disease and asthma.70 LTB4 is rapidly expressed by phagocytic cells (PMNs and macrophages) following stimulation with bacterial LPS or fMLP. More recently, the LTB4 receptor, BLT1, has been implicated in preferential recruitment of Th2 type T lymphocytes during allergic pulmonary responses.71–73 This receptor has also been implicated in recruitment of T lymphocytes that mediate lung allograft rejection and development of obliterative bronchiolitis in lung allographs in rodents.74 Thus, LTB4 and BLT1 are now being considered as targets for therapy in chronic immune responses in the lung, in contrast to their traditional roles as potent neutrophil and mononuclear cell chemoattractant in acute lung injury.
Chemokines and Immune Cell Migration
Chemokines (discussed in chapter 26) have been divided into two main families based upon their sequence homology and the position of the first two cysteine residues, C-x-C (alpha) and C-C (beta).75,76 There are two minor families described each with a single member, the C and Cx3C families. Much of our understanding of chemokines has centered upon their role in mediating leukocyte recruitment to the site of inflammation in lung or specifically directing recirculation of leukocytes during homeostasis. Interestingly, results have indicated that many of these family members also have diverse roles in the activation and differentiation of various immune and nonimmune cell populations. While the chemokine family members’ function is diverse, the promiscuous binding relationship between multiple members with a single receptor as well as a specific receptor being able to bind multiple chemokines underscores our relative lack of understanding of the biology of the chemokine family. In particular, it is often difficult to understand how such a vast number of chemotactic molecules, several being produced simultaneously, could coordinate inflammatory responses. Recent studies demonstrate that it may be the overall profile of chemokines being produced that dictates the inflammatory cell response resulting in leukocyte accumulates at a site of injury or infection. This latter aspect can be best displayed during acute inflammatory responses, such as in bacterial infections, when the cellular infiltrate is primarily neutrophilic.77 Chemokines that bind to CxCR1 and CxCR2 mediates this process. Likewise, when more insidious pathogens are present and acute inflammatory mechanisms cannot effectively control the infectious process, immune cytokines, such as IFN and IL-4, tend to drive the production of chemokines that allow the mononuclear cells, macrophages, and lymphocytes to accumulate at sites of infection, resulting in a more effective immune response for enhanced clearance of the pathogen. Thus, although there are numerous chemokines being produced during any single inflammatory response, the overall profile of the response may be directed to recruitment of cells that are most appropriate to deal with particular stimuli. These “fine tuned” responses mediated by chemokines also depend upon the chemokine receptor profile of the transmigrating leukocyte (Fig. 23-2). For example, while many leukocytes such as PMNs and macrophages tend to have a fairly fixed chemokine receptor expression (CxCR2 for PMNS and CCR2 for inflammatory macrophages),78–80 T cell subsets (Th1, Th2, Th17, Treg cells) express a differential profile of chemokine receptors that may preferentially allow recruitment to specific types of inflammatory/immune responses.81–84 Those aspects will be discussed in later sections.
LEUKOCYTE ACCUMULATION AND LUNG PATHOLOGY
A variety of leukocytes may accumulate with the lung. Each is discussed below.
 NEUTROPHILS
NEUTROPHILS
The accumulation of neutrophils (PMNs) in the lung is the first line of defense against infectious organisms. There are significant numbers of PMNs circulating normally, and they can be quickly mobilized from the bone marrow during inflammatory responses or during acute lung injury. Once activated at the site of inflammation, PMNs preform phagocytic and bacterial/fungal killing functions that promote clearance of bacterial and fungal pathogens. They also can quickly release a number of enzymes, (proteases, etc.), that can have detrimental effects on the local lung tissue and cause physiologic dysfunction and severe damage if not tightly regulated. Individuals with one of many abnormalities in PMN formation, recruitment, or activation defects often develop recurrent, severe infections, both bacterial and fungal.85,86 As described earlier, PMNs enter the lung via an initial adhesion event that progresses via a selectin-mediated rolling and subsequently through a β2-integrin (CD11b/CD18)/ICAM-1–induced firm adhesion. While controversial evidence using animals exists, it appears that entry into the lung via the alveolar vasculature may not require the entire adhesion progression since there is low shear stress, allowing leukocytes to migrate into the airspace with less resistance.87–90 This is because the diameter of the neutrophil and the capillary is nearly the same, which would not allow rolling. Nevertheless, in the alveolar vasculature the adhesion molecules described in section II are still present, functional, and are required for ultimate PMN transmigration. Not surprisingly, numerous chemotactic factors can quickly mobilize and mediate PMN migration into inflamed tissues. These factors include bacterial products such as fMLP, an N-formylated 3 amino acid peptide that is produced by numerous bacteria. fMLP binds to a GPCR (fMLPR). Other chemotactic factors expressed by the host early during pathogen responses include C3a and C5a, as well as the primary lipid mediator, LTB4, all interacting with specific GPCR signaling on the surface of PMNs. Usually, the early migration of PMNs to the lung is likely due to the latter mediators that are quickly cleaved or stored in cells (such as in mast cell granules) and are readily activated or released upon pathogenic or injurious stimuli. Subsequently, the prolonged activation of the pulmonary environment beyond immediate responses (>4 hours) results in cytokine cascades leading to additional and numerous more efficient chemokine protein mediators of migration, including CxCL1 (GROα), CxCL5 (ENA-78), and CxCL8 (IL-8). The CxC family chemokines are produced by both immune and nonimmune cell populations and provide relative specificity for PMN accumulation during lung inflammatory disease via their cognate receptors, CxCR1 and CxCR2, predominantly found on PMNs. Increased numbers of PMNs are often found in severe inflammatory diseases of the lung and likely provide nonspecific damage that may result in lung dysfunction. Several strategies for blocking adhesion as well as chemotactic receptors are currently under development by numerous pharmaceutical companies.
 EOSINOPHILS
EOSINOPHILS
The role of eosinophils evolutionarily has been linked to chronic parasitic diseases, in which they perform a protective killing response linked to parasite clearance.91,92 However, as the parasitic burden in humans has been greatly reduced, the immune responses, especially at mucosal surfaces, have led to detrimental chronic responses to inert parasitic antigens. This has led to a number of allergic inflammatory diseases, especially allergic asthma in the lung. Eosinophils are derived in the bone marrow, primarily in the presence of a Th2 type response that provides systemic IL-5 levels that feedback to the bone marrow, directing the maturation and release of eosinophils into circulation. The migration of eosinophils into the lung and other tissues rely on a different subset of adhesion and chemotactic factors for entry into the tissue compartments.93–99 While the migratory adhesion pathway has not been as thoroughly defined, it appears that it also relies on an initial combination of selectin and β-integrin–mediated adhesion events prior to responding to chemotactic mediators in the lung. While eosinophils are able to utilize CD11b/CD18-ICAM-1–mediated migration pathways, it appears that more efficient and preferential migration is VLA-4 β-integrin–mediated VCAM-1 adhesion. Since VCAM-1 is highly upregulated in the lung during Th2 cytokine–mediated responses, as in asthma, such responses may relate to the inflammatory cytokine environment that stimulates maturation of eosinophils in the bone marrow (IL-5 mediated). Subsequent to adhesion of eosinophils to endothelial cells, eosinophil migration and accumulation in lung can also be regulated by the chemotactic factors that appear to preferentially recruit eosinophils. While C5a and the lipid mediators, LTB4 and PAF, can each provide a stimulus for such migration, it appears that chemokines are the primary stimuli for migration of eosinophils. In particular, CCR3 ligands are the most potent, including CCL5 (RANTES), CCL11 (eotaxin-1), CCL24 (eotaxin-2), and CCL26 (eotaxin-3). CCR3 is the characteristic chemokine receptor expressed on eosinophils and although they also appear to express CCR1, it appears to have only a minor role in the recruitment during chronic disease. Similar to the PMN, eosinophils can induce local damage in lung after their degranulation and release of proteases and enzymes. In addition, eosinophils have been implicated in progression of remodeling diseases, linked to their interaction with fibroblasts.100,101 Eosinophils have the ability to transform normal fibroblasts into matrix-producing myofibroblasts. Eosinophils have been identified as a significant source of TGFβ, FGF, as well as other pro-fibrotic factors and have been implicated in severe remodeling in chronic allergic and inflammatory diseases.102,103 Thus, significant effort continues to be made to target eosinophil migration during chronic pulmonary diseases, especially asthma.
 MONOCYTES/MACROPHAGES
MONOCYTES/MACROPHAGES
Resident macrophage populations play an important role in the lung, providing initial protection against pathogenic and noxious damage. The alveolar and interstitial macrophages that reside in and around airways appear to be ideally suited, having optimal ability to phagocytize and kill microorganisms as well as producing regulated levels of inflammatory cytokines. Macrophages also function for uptake of inhaled particles. Phagocytosis requires an intact cytoskeleton and is most efficient when phagocytosis is mediated by Fc receptors. Complement and scavenger receptors such as MARCO are also important mechanisms for mediating clearance of microorganisms.104–107 During responses to infectious and inflammatory stimuli, the migration of monocytes from the blood can also play an important role in the clearance of the inciting agents. In humans, there appear to be two distinct subsets of circulating monocytes, CD14+ monocytes with high CCR1, CCR2 and CxCR2 expression, and low Cx3CR1 expression as well as a distinct CD16+ population of monocytes with high levels of Cx3CR1 and low levels of CCR2 expression.108–110 Similar subsets exist in mice and, while not exact, this allows characterization of their different roles in disease once recruited to the site of inflammation allowing extrapolation to human disease. Mice lacking inflammatory, CCR2+ monocytes are highly susceptible to Listeria monocytogenes and Mycobacterium tuberculosis infections, demonstrating that infiltrating monocytes are important effector cells for the clearance of intracellular bacteria.111,112 In a similar fashion the inhibition of inflammatory macrophage infiltration into the lung during Aspergillus fumigatus and Cryptococcus infections can lead to prolonged and detrimental infection by these organisms.113,114
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


