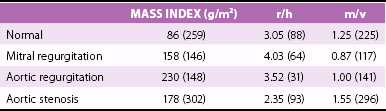
Chapter 5 In 1973, Grossman et al 1 proposed the schema shown in Figure 5-1 as the foundation on which the left ventricle responds to valvular heart disease. 1 In this concept, the increased systolic stress (σ) caused by pressure overload induces sarcomere production in parallel, increasing myocyte width, and in turn increasing left ventricular (LV) wall thickness. Because σ = p × r/2h, where p = LV pressure, r = LV radius, and h = thickness, increased pressure in the numerator is offset by increased thickness in the denominator so that stress remains normal. Systolic wall stress is a reasonable surrogate for LV afterload. Because the ejection fraction varies inversely with afterload, the concentric hypertrophy and remodeling that occur through this process are thought of as initially compensatory because they help maintain LV function. FIGURE 5-1 Effect of pressure and volume overload on development of left ventricular hypertrophy. In the same hypothesis, volume overload increases diastolic stress that causes sarcomeres to be laid down in series, lengthening each myocyte and in turn increasing LV volume. Greater volume then allows total stroke volume to increase, helping to compensate for the wasted volume lost to regurgitation. Because this mechanism is a requisite to normalizing forward stroke volume, it too is considered compensatory, at least in part. Table 5-1 demonstrates the patterns of remodeling found in the left-sided overloading valve lesions. 2 AS, the classic pressure overload lesion, produces typical concentric LV hypertrophy (LVH) with the highest mass : volume (m/v) ratio and the lowest radius : thickness (r/h) ratio. MR is the prototypical volume overload lesion, leading to the lowest mass : volume ratio and the highest radius : thickness ratio. AR, a combined LV pressure and volume overload, causes the greatest amount of LVH with hybrid geometry between that of AS and MR. TABLE 5-1 Hypertrophy in Human Left-Sided Overload Valve Lesions * m/v, ratio of left ventricular mass to volume; r/h, ratio of left ventricular radius to thickness. *Numbers in parentheses indicate numbers of subjects analyzed. From Carabello BA. The relationship of left ventricular geometry and hypertrophy to left ventricular function in valvular heart disease. J heart Valve Dis 1995(Suppl 2):S132–8. It has become fashionable to use the term remodeling when discussing changes in ventricular size and geometry, and most remodeling is associated with LVH. However, the two terms are not synonymous. Hypertrophy means an increase in mass, whereas remodeling indicates a change in geometry and/or volume. Thus, a situation in which increased wall thickness is accompanied by increased LV mass should be considered concentric hypertrophy, whereas one in which increased thickness is accompanied by a reduction in volume, leading to no change in LV mass, 3 should be termed concentric remodeling. Although the term remodeling is most often used in the context of LV dilation, a distinction should also be made between ventricles that enlarge with concomitant wall thinning, in which LV mass could remain the same (pure remodeling), and left ventricles that also increase their mass (eccentric hypertrophy) ( Figure 5-2). The normal aortic valve opens slightly, through not entirely understood mechanisms, before pressure in the left ventricle exceeds aortic pressure4,5 and then offers almost no resistance to LV outflow. As the valve becomes diseased it stiffens and the orifice area diminishes. Even when aortic valve area is reduced by half, the LV pressure exceeds that of the aorta by only 5 to 10 mm Hg. However, further reductions in aortic valve area cause progressively greater pressure gradients across the valve. The transvalvular gradient represents the additional pressure that the left ventricle must generate to drive blood past the obstruction to outflow. It is generally agreed that this mechanical stress is transduced into a biological response, leading to hypertrophy and/or remodeling. As noted previously, because the development of hypertrophy helps normalize afterload, thereby normalizing ejection performance, such hypertrophy has been viewed as compensatory. According to the paradigm proposed in Figure 5-1, just enough hypertrophy should develop to return wall stress to normal. Indeed, in some cases this expected course is borne out. However, such perfect compensation often fails to occur ( Figures 5-3 and 5-4). Figure 5-3 demonstrates that frequently in patients with LV dysfunction, such dysfunction is due to afterload excess, indicating that not enough hypertrophy developed to normalize stress. 6 In fact, the majority of patients with AS have some element of afterload excess, indicating a lack of fully compensatory hypertrophy. 7 FIGURE 5-3 Wall stress and left ventricular systolic function in aortic stenosis. FIGURE 5-4 Wall stress and left ventricular systolic function in aortic stenosis. The opposite end of the spectrum is shown in Figure 5-4. 8 In some patients, especially elderly women and children born with congenital AS, 9 there appears to be excessive hypertrophy. In such patients afterload is actually subnormal, leading to higher than expected ejection performance, at least at the endocardial level. It should be noted that assessment of LV function at the endocardial level often overestimates contractility. Ejection of blood from the LV cavity during systole is primarily a function of wall thickening. The more sarcomeres present in parallel, the more thickening occurs with shortening of the sarcomeres. Thus, in concentric remodeling and hypertrophy, subnormal shortening can still produce a normal ejection fraction. 10 Therefore, for accurate assessment of LV function in concentrically altered ventricles, midwall shortening should be evaluated; and when done, this evaluation may reveal diminished LV function, although not always. In still other patients yet another response—concentric remodeling—develops to AS. In these patients there is no increase in LV mass. 3 Rather there is a reduction in LV volume together with an increase in LV wall thickness, acting to normalize stress without actual hypertrophy. The question arises, why is there such inhomogeneity in the hypertrophic response to pressure overload? Is the differing LV geometry that occurs a response to different disease characteristics, such as valve area, rate of progression, or body habitus? Or is there an inherent difference in response to a similar pressure overload? Koide et al 11 addressed this question by creating a model of AS in which a gradually imposed gradient was identical in dogs of similar size and weight. The hypertrophy that subsequently developed recapitulated that seen in humans. Some animals had modest concentric hypertrophy, whereas others had severe hypertrophy. Of interest, the group with modest hypertrophy had persistently higher wall stress yet far less myocardial mass despite this greater stimulus for hypertrophy ( Figure 5-5). These data suggest a different set point for response to the overload for which the stimulus was greater and the response less. It is likely that these inherent differences also explain the difference in the hypertrophic response noted in humans. FIGURE 5-5 Heterogeneous response to pressure overload. LV ejection is controlled by preload, afterload, and contractility. The reduction in the ejection fraction in AS stems from increased afterload, decreased contractility, or both.7,12 Increased afterload occurs when remodeling fails to offset the greater systolic pressure required of the left ventricle. Whereas concentric LVH has long been considered a compensatory mechanism, this issue is not clear-cut. In studies of hypertrophy in general, LVH has led to increased cardiac mortality, especially in the presence of coronary artery disease. 13 Yet genetic maneuvers in mice that prevent or diminish the hypertrophic response have led to both increased mortality14,15 and, conversely, beneficial effects,16,17 leaving the question of the compensatory role of LVH in doubt. In the canine model already described, contractility was preserved at both sarcomere and LV chamber levels in the animals with extreme hypertrophy in which wall stress was normalized. In dogs with high afterload, contractility was depressed, at least in part owing to microtubular hyperpolymerization, which acted as an internal stent inhibiting sarcomere shortening. 18 Conversely, in a human study of AS, the best outcome was in patients without LVH who underwent concentric remodeling. 3 LV mass was not greater, but increased relative wall thickness normalized afterload, allowing for compensated systolic function. This observation is not universal, however. In some patients the concentrically hypertrophied left ventricle is so reduced in volume that stroke volume is decreased. In turn, a small stroke volume reduces the transaortic gradient, potentially misleading the clinician into underestimating disease severity, thereby delaying therapy and leading to increased surgical risk. 19 In still another study, Duncan et al 20 propensity matched 964 pairs of patients with AS undergoing aortic valve replacement with and without concentric LVH and/or remodeling. Patients with concentric LVH had double the operative risk and double the postoperative morbidity in comparison with patients without this pattern. 20 It seems likely that intrinsic LV biological factors beyond the simple imaged geometry and mass of the LV account for these disparate results regarding the compensatory versus deleterious effects of LVH. Our future ability to understand and detect these factors should give us a better understanding of the role of LVH in AS. When contractile dysfunction does occur, its mechanism is probably multifactorial. Concentric LVH clearly results in abnormal coronary blood flow and blood flow reserve.21–23 Normally the subendocardium receives about 20% more blood flow than the epicardium, but this ratio is reversed in LVH. 24 Thus, the myocardial layer with the highest oxygen demand receives the least oxygen supply. Further, coronary reserve is limited in concentric LVH. Whereas in normal individuals, coronary flow can increase by fivefold to eightfold in response to increased myocardial energy demands, flow reserve in AS is limited to an increase of twofold to threefold. 21 Abnormal flow reserve and flow distribution lead to subendocardial ischemia and contractile dysfunction during periods of stress. 24 It is also possible, but unproven, that this chronic imbalance could cause myocardial hibernation or stunning. The cytoskeletal abnormalities noted previously as well as disordered calcium handling and apoptosis also probably play a role.25–27 Finally, there is the general belief that LVH transitions from a compensatory phase to a pathologic one. 28 Although certainly plausible, this concept, too, has been questioned. Animals destined to have contractile dysfunction demonstrated gene expression different from that in those who maintained normal function early in the course of pressure overload, suggesting that two separate patterns of hypertrophy exist rather than one that transitions into another, 29 consistent with the Koide dog model. It is well recognized that diastolic function is abnormal in concentric LVH. Dysfunction accrues from delayed relaxation, increased wall thickness, and changes in myocardial structure with an increase in stiffness mediated by greater collagen content.30,31
Left Ventricular Adaptation to Pressure and/or Volume Overload
 Key Points
Key Points
 Each valve lesion imparts a unique hemodynamic load on the left ventricle, wherein aortic stenosis creates a pure pressure overload, mitral regurgitation presents a pure volume overload, aortic regurgitation causes combined pressure and volume overload, and mitral stenosis leads to volume underload and potentially increased afterload. In turn, each lesion causes its own type of hypertrophy and remodeling.
Each valve lesion imparts a unique hemodynamic load on the left ventricle, wherein aortic stenosis creates a pure pressure overload, mitral regurgitation presents a pure volume overload, aortic regurgitation causes combined pressure and volume overload, and mitral stenosis leads to volume underload and potentially increased afterload. In turn, each lesion causes its own type of hypertrophy and remodeling.
 The terms remodeling and hypertrophy are not synonymous.
The terms remodeling and hypertrophy are not synonymous.
 In almost all cases hypertrophy is both adaptive and maladaptive.
In almost all cases hypertrophy is both adaptive and maladaptive.
Background

The diagram shown is a framework for how mechanical stress (σ) is transduced into pressure versus volume overload hypertrophy. (From Grossman W, Jones D, McLaurin LP. Wall stress and patterns of hypertrophy in the human left ventricle. J Clin Invest 1975;53:332–41.)
Hypertrophy versus Remodeling
Aortic Stenosis
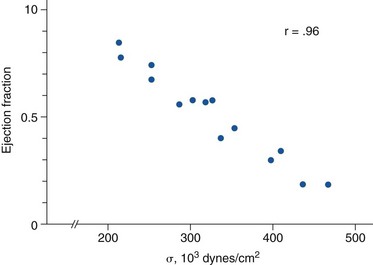
Ejection fraction is plotted against mean systolic wall stress (afterload; σ) for patients with aortic stenosis. As afterload increases from inadequate left ventricular hypertrophy, ejection fraction falls. (From Gunther S, Grossman W. Determinants of ventricular function in pressure-overload hypertrophy in man. Circulation 1979;59:679–88.)
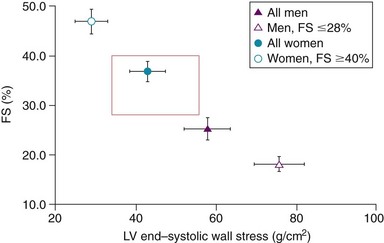
Fractional shortening (FS) is plotted against left ventricular (LV) systolic wall stress for patients with aortic stenosis. Some patients, especially women, have abnormally low stress and very high shortening fractions, suggesting that more left LV hypertrophy is present than that needed simply to normalize stress. (From Carroll JD, Carroll EP, Feldman T, et al. Sex-associated differences in left ventricular function in aortic stenosis of the elderly. Circulation 1992;86:1099–107.)
Variability in the Response to Pressure Overload
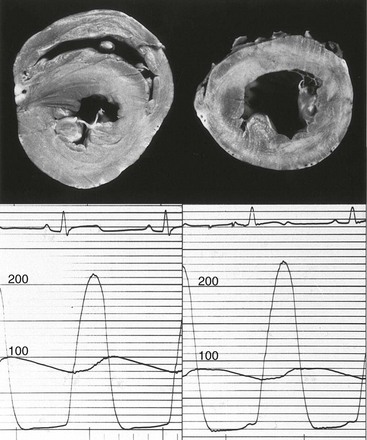
Left ventricles from two dogs (top) with identical gradients (bottom) demonstrate different degrees of hypertrophy. (From Koide M, Nagatsu M, Zile MR, et al. Premorbid determinants of left ventricular dysfunction in a novel mode of gradually induced pressure overload in the adult canine. Circulation 1997;95:1349–51.)
Concentric Hypertrophy and Left Ventricular Function
![]()
Stay updated, free articles. Join our Telegram channel




Full access? Get Clinical Tree


