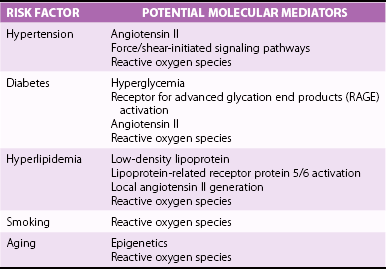
Chapter 3 GENERAL CONCEPTS AND HISTOLOGIC CHANGES RISK FACTORS CONVERGE ON A PATHOGENIC RESPONSE TO CHRONIC INJURY CLINICAL OBSERVATIONS IN HUMANS INDUCTION OF OSTEOGENIC SIGNALING CASCADES MODULATORS OF OSTEOGENIC DIFFERENTIATION Direct Inhibitors of Osteogenic Signaling Inflammatory Cell Infiltrate and Proinflammatory Cytokines Peroxisome Proliferator-Activated Receptor γ Signaling FIBROSIS AND MATRIX MODULATION OF CALCIFICATION MOLECULAR PATHWAYS CONTRIBUTING TO NONOSTEOGENIC CALCIFICATION TRANSLATION OF BIOLOGICAL FINDINGS TO THERAPEUTIC INTERVENTIONS Fibrocalcific aortic valve disease was once thought to be a degenerative disease characterized by the passive accumulation of calcium in the valve cusp. Landmark studies over the past decade, however, demonstrated that osteoblast and osteoclast markers are frequently on cells in valves from patients with this disease, and subsets of patients have evidence of bone matrix in the calcified regions of the valve. 1 Collectively, these observations suggest that aortic valve calcification and expansion of calcified deposits constitute an active process similar to that observed in bone. The ensuing sections focus on cellular and molecular mechanisms regulating the formation and activity of osteoblast-like cells in the calcifying aortic valve and propose potential therapeutic strategies to slow or reverse progression of calcific valve disease. The normal aortic valve comprises three layers: the fibrosa, spongiosa, and ventricularis. 2 Importantly, each layer contains valvular interstitial cells, which play a critical role in the production, maintenance, and repair of each layer ( Figure 3-1). 3 Both the aortic and ventricular sides of the valve are covered by an endothelial monolayer ( Table 3-1). FIGURE 3-1 Key histologic and structural characteristics of the valve and changes during initiation and progression of valve disease. Disruption of the aortic valve endothelium is thought to be one of the earliest histopathologic changes contributing to the initiation of fibrocalcific aortic valve disease in humans and experimental animals (see Figure 3-1). 4 This is typically accompanied by infiltration of T lymphocytes and macrophages.4–7 The subsequent elaboration of profibrotic and inflammatory cytokines elicits increased extracellular matrix production and turnover, finally resulting in stiffening and fibrosis of the extracellular matrix in the early valve lesions. 8 Early valve lesions may also exhibit lipid accumulation of extracellular lipid, which is ultimately taken up by macrophages to become foam cells. Calcium deposition is commonly observed in early stages of fibrocalcific aortic valve disease and frequently co-localizes with areas showing inflammatory cell infiltration.4,7 During advanced stages of fibrocalcific valve disease there is massive accumulation of lipid and inflammatory cell infiltrate, as well as increased production of a disorganized, stiffened extracellular matrix and fragmented elastic lamina.4,5,8 Extensive cusp calcification, however, is the hallmark histopathologic change associated with hemodynamically significant aortic valve stenosis. 4 As discussed in more detail later, accumulation of calcium can appear either amorphous (i.e., no clear organization/bone structure) or osteogenic (i.e., evidence of endochondral ossification, bone matrix, and hematopoietic marrow compartments).1,4,5,9 Risk factors for development of aortic valve disease are remarkably similar to those for atherosclerosis, and include increasing age, hypercholesterolemia, hypertension, smoking, and diabetes.10,11 (See Figure 3-2 and Chapter 4.) The observation that the phenotypic penetrance of any one risk factor is highly variable suggests that there is a complex interplay between the environment and the genome (i.e., genetic propensity to mount an excessive response to injury) ( Table 3-2). In the context of fibrocalcific aortic valve stenosis, this pathophysiologic response results in massive accumulation of calcium in the valve cusp. TABLE 3-2 Risk Factors for Development of Fibrocalcific Aortic Valve Disease and Their Potential Molecular Mediators FIGURE 3-2 Schematic working model of the interplay between risk factors for fibrocalcific aortic valve disease and the dysfunctional “response to injury” that can increase propensity for development of valvular stenosis. In 2001, Mohler et al 1 reported that approximately 20% of aortic valves from humans with severe aortic stenosis had evidence of bone matrix at the time of valve surgery. Histologically, the bone matrix was associated with the presence of cells that resembled osteoblasts and osteoclasts ( Figure 3-3). These findings were particularly remarkable given the fact that aortic valve calcification was typically viewed as a passive, degenerative process by clinicians and scientists alike, and they represented a paradigm shift in the field of fibrocalcific aortic valve disease. FIGURE 3-3 Histopathologic evidence of osteochondrogenic changes in fibrocalcific aortic valve disease. More recently, the role of fibrosis and matrix remodeling in the pathogenesis of aortic valve calcification and dysfunction has received a substantial amount of attention. Massive valvular calcification is nearly ubiquitously associated with increases in extracellular matrix accumulation and turnover.12–14 While this was once thought to be an epiphenomenon, an emerging body of experimental data suggests that changes in the extracellular matrix of the valve may not only be a major modulator of valve interstitial cell function, but may also be sufficient to impair valvular dysfunction in advanced fibrocalcific aortic valve disease. Although description of cellular and molecular changes in human tissue is a critical component in research, empirical testing in animal models is an essential step in discerning whether a change is a pathophysiologic driver of fibrocalcific aortic valve disease or merely an epiphenomenon ( Figure 3-4). When one is evaluating experimental data or deciding which model is useful for a particular study design, several important questions must be asked ( Table 3-3): TABLE 3-3 Echocardiographic and Hemodynamic Changes in Animal Models of Aortic Valve Sclerosis and Stenosis FIGURE 3-4 Methods to evaluate aortic valve function in mice. • Does the model require genetically altered animals, and do the mutations relate to the specific question at hand? • What is the underlying stimulus driving calcification (e.g., hyperlipidemia, hypertension)? • Are the histopathologic changes relevant to human disease (e.g., fibrosis and calcification)? • Do the animals develop hemodynamically significant aortic valve dysfunction and stenosis, or only aortic valve sclerosis? Numerous studies have reported increased expression of multiple bone morphogenetic protein (BMP) isoforms in diseased human valves, including BMP2, BMP4, and BMP6.14–20 In general, BMP elaboration is thought to originate from the endothelium on the aortic face of the valve 21 ( Figure 3-5), where shear forces are nonlaminar and inhibitors of BMP signaling are disproportionately low. 15 Binding of BMPs to their receptor complex on aortic valve interstitial cells results in Smad1/5/8 phosphorylation and subsequent translocation of the Smad complex to the nucleus, where it drives pro-osteogenic gene expression through its binding to Smad binding elements.22,23 Although Smad6 appears to play a major role in tonic suppression of BMP signaling (because Smad6-null mice have evidence of cardiovascular calcification at 2 weeks of age 24), the role of other inhibitory molecules in the regulation of aortic valve calcification (e.g., Smurf1/2) remains poorly understood. FIGURE 3-5 Changes in bone morphogenetic protein signaling in fibrocalcific aortic valve disease. Bone morphogenetic protein signaling is also elevated in experimental animal models of fibrocalcific aortic valve disease. Importantly, increases in Smad1/5/8 phosphorylation precede aortic valve dysfunction in hypercholesterolemic mice, 6 suggesting that increases in BMP signaling are not simply an epiphenomenon associated with end-stage valve calcification and stenosis. A second major osteogenic pathway activated in fibrocalcific aortic valve disease is Wnt/β-catenin signaling ( Figure 3-6). In brief, activation of the canonical signaling pathway involves binding of Wnt ligands to a receptor complex that results in activation and nuclear translocation of β-catenin, which can subsequently drive pro-osteogenic gene expression.25,26 Multiple components of this pathway have been implicated in calcified human and animal aortic valves, including Wnt ligands (Wnt3a, Wnt7a),27–30 lipoprotein receptor–related protein (lrp) receptor complex components (LRP5/6, frizzled receptors),29,31 and nuclear translocation of the β-catenin transcription factor complex.28,30–32 FIGURE 3-6 Changes in Wnt/β-catenin signaling in experimental animal models of fibrocalcific aortic valve disease. Wnt/β-catenin signaling can be negatively regulated at multiple levels, including through inhibition of Wnt binding, inhibition of β-catenin activation, and proteasomal degradation of β-catenin.25,26 The role of changes in endogenous inhibitors of the Wnt/β-catenin signaling pathway in the pathogenesis of fibrocalcific aortic valve disease remains poorly understood. Like BMP signaling, canonical tumor growth factor-β (TGF-β) signaling involves phosphorylation of Smad proteins (Smad2/3 in particular) and translocation of the activated Smad complex to the nucleus.22,33 Increases in TGF-β expression, Smad2/3 phosphorylation, and multiple Smad2/3 target genes have been shown in humans and animals with fibrocalcific aortic valve disease6,32,34–37 ( Figure 3-7). FIGURE 3-7 Role of tumor growth factor-β (TGF-β) signaling in fibrocalcific aortic valve disease. The role of canonical TGF-β signaling in the initiation and progression of calcification in aortic valve disease, however, is controversial. The key observation suggesting that TGF-β contributes to calcification in valve disease is that cultured aortic valve interstitial cells treated with exogenous TGF-β rapidly form calcified nodules via a caspase/apoptosis-dependent mechanism.35,38 There are, however, several observations from in vivo model systems suggesting that TGF-β may not accelerate valve calcification in fibrocalcific aortic valve disease. First, lipid lowering in mice with advanced aortic valve disease reduces osteogenic gene expression and does not reduce Smad2/3 phosphorylation, 32 suggesting that TGF-β is not a primary driver of osteogenic gene expression in fibrocalcific aortic valve disease. Second, mice that are deficient in one copy of Smad3 (i.e., Smad3+/− mice) have a higher bone mineral density than their wild-type littermates, 39 suggesting that TGF-β may suppress osteogenesis in bone. Finally, although canonical TGF-β signaling may not promote (or may even inhibit) valve calcification, emerging data suggest that TGF-β receptor activation may transactivate Wnt/β-catenin signaling, 30 which is likely to promote interstitial cell osteogenesis. Future studies with experimental manipulation of TGF-β signaling in robust, in vivo models of fibrocalcific aortic valve disease will be essential to define its role in valve calcification and stenosis. Reductions in expression of inhibitors of osteogenic signaling are likely to play a significant role in initiation and progression of fibrocalcific aortic valve disease. As mentioned previously, genetic deletion of Smad6 results in cardiovascular calcification in the absence of additional exogenous stressors, 24 suggesting that tonic suppression of BMP signaling is critical for prevention of valvular calcification. It would also appear that tonic BMP ligand sequestration/neutralization is important in preventing cardiovascular calcification because mice deficient in matrix Gla protein (which binds and inactivates BMP2) demonstrate spontaneous cardiovascular calcification early in life, 40 and mice overexpressing matrix Gla protein are protected against hypercholesterolemia-induced vascular calcification. 41 Although matrix Gla protein levels are under transcriptional and translational regulation, the posttranslational gamma-carboxylation of matrix Gla protein is required for binding to BMP2.42–46 Clinically, several retrospective studies reported that drugs that inhibit gamma-carboxylase (e.g., warfarin) have been associated with increased risk of cardiovascular calcification and aortic valve stenosis.47–50 Along these lines, administration of warfarin to juvenile rats results in significant vascular calcification.51–55 Collectively, tonic suppression of BMP signaling, at both intracellular and extracellular levels, appears to be important in prevention of cardiovascular calcification. Hypertension is a major risk factor for development of fibrocalcific aortic valve disease, and is frequently associated with systemic increases in activity of the renin-angiotensin system (RAS). It is often underappreciated, however, that the “local” RAS can be a major contributor to rises in tissue levels of angiotensin II. A large amount of work has shown that increases in tissue RAS activity are major contributors to inflammation, oxidative stress, fibrosis, and plaque expansion in atherosclerotic lesions.56–58 In stenotic aortic valves, infiltrating macrophages are abundant and can be primary sources of greater local RAS activity and concomitant rises in tissue angiotensin II levels6,7,59–61 ( Figure 3-8). Interestingly, increases in chymase activity can also convert angiotensin I to angiotensin II, and macrophages express high levels of chymase.58,59 Retrospective studies suggest that angiotensin-converting enzyme (ACE) inhibition may slow progression of aortic valve disease, 62 there are limited experimental data testing this hypothesis, and the fact that chymase activity is unaffected by ACE inhibitors makes it a less appealing target. Preclinical experiments in hypercholesterolemic rabbits, however, showed that angiotensin 1 receptor blockade attenuates aortic valve interstitial cell activation, endothelial disruption, and valvular inflammation in early stages of valve disease, 63suggesting that angiotensin II and angiotensin 1 receptor activation may be important factors even in early stage fibrocalcific aortic valve disease. FIGURE 3-8 Changes in angiotensin II (ANGII)–related molecules in fibrocalcific aortic valve disease. Inflammatory cells such as macrophages, 64 neutrophils,65,66 and T cells 67 are ubiquitous findings in end-stage human aortic valve disease and in most animal models of fibrocalcific valve disease. Consistent with this finding, elaboration of proinflammatory cytokines—for example, tumor necrosis factor-α (TNFα) and interleukins IL-6 and IL-1—is also dramatically increased in human and animals with fibrocalcific aortic valve disease.68–72 Although few studies have examined the role of proinflammatory cytokines in the progression of fibrocalcific aortic valve disease, three lines of evidence suggest that TNFα may play a central role in the initiation and progression of disease. First, aortic valves from interleukin-1 receptor antagonist (IL-1ra)–deficient mice are thickened, accumulate calcium, and develop mild aortic valve dysfunction (peak transvalvular velocity 2 m/sec) 68 ( Figure 3-9). Importantly, this phenotype is abolished in IL-1ra/TNFα double-knockout mice, suggesting that TNFα is the major downstream mediator of IL-1–induced inflammation. 68 FIGURE 3-9 Role of proinflammatory signaling in the pathogenesis of fibrocalcific aortic valve disease.
Cellular and Molecular Basis of Calcific Aortic Valve Disease
General Concepts and Histologic Changes
General Concepts
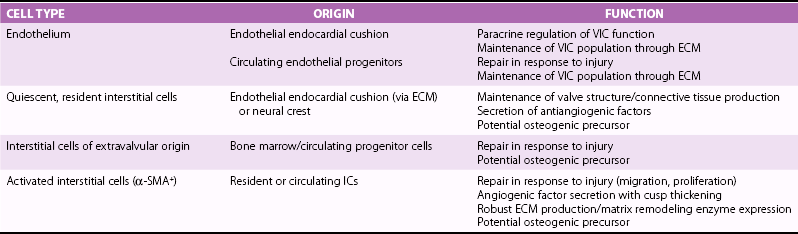
Key Histologic Changes During Initiation and Progression of Valve Disease
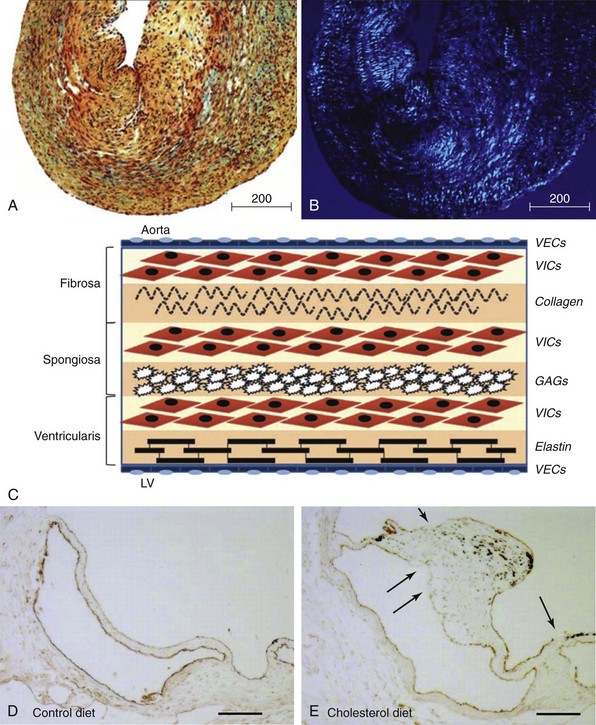
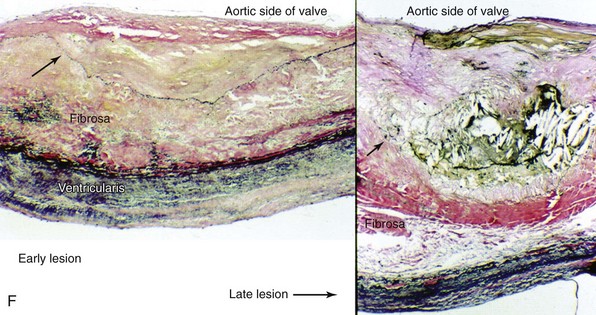
Movat’s pentachrome staining (A) and polarized light (B) imaging of a normal porcine aortic valve cusp depicting the trilayered structure of the aortic valve. Note that the matricellular composition varies throughout the valve. In A, black indicates nuclei and elastic fibers, yellow collagen, blue mucin, bright red fibrin, and dark red muscle. C, Schematic depicting the different layers of the aortic valve cusp (layering/orientation is the same as in A and B). D and E, Micrographs depicting disruption of the aortic valve endothelium (arrows) in mice fed a western-type diet for 16 weeks (immunohistochemical staining using anti–endothelial nitric oxide synthase antibody). F, Micrographs depicting cusp thickening in early stages of fibrocalcific aortic valve disease (left) and a massive calcific deposit in an aortic valve cusp during later stages of fibrocalcific aortic valve disease (right). Note that calcification and matrix disruption predominantly affect the aortic side of the valve. GAGs, Glycosaminoglycans; LV, left ventricle; VECs, vascular endothelial cells; VICs, valve interstitial cells. (A and B from Simionescu DT, Chen J, Jaeggli M, et al. Form follows function: advances in trilayered structure replication for aortic heart valve tissue engineering. J Healthc Eng 2012;3:179–202; C from Leopold JA. Cellular mechanisms of aortic valve calcification. Circ Cardiovasc Interv 2012;5:605–14; D and E from Matsumoto Y, Adams V, Jacob S, et al. Regular exercise training prevents aortic valve disease in low-density lipoprotein-receptor-deficient mice. Circulation 2010;121:759–67; F from Freeman RV, Otto CM. Spectrum of calcific aortic valve disease: pathogenesis, disease progression, and treatment strategies. Circulation 2005;111:3316–26.)
Risk Factors Converge on a Pathogenic Response to Chronic Injury
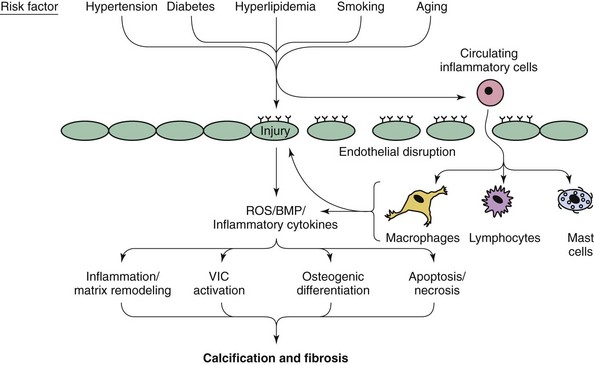
Note that both valvular calcification and fibrosis are likely to play major roles in the development of hemodynamically significant aortic valve dysfunction. BMP, Bone morphogenic protein; ROS, reactive oxygen species; VIC, valvular interstitial cell. (Modified from Miller JD, Weiss RM, Heistad DD. Calcific aortic valve stenosis: methods, models, and mechanisms. Circ Res 2011;108:1392–412.)
Clinical Observations in Humans
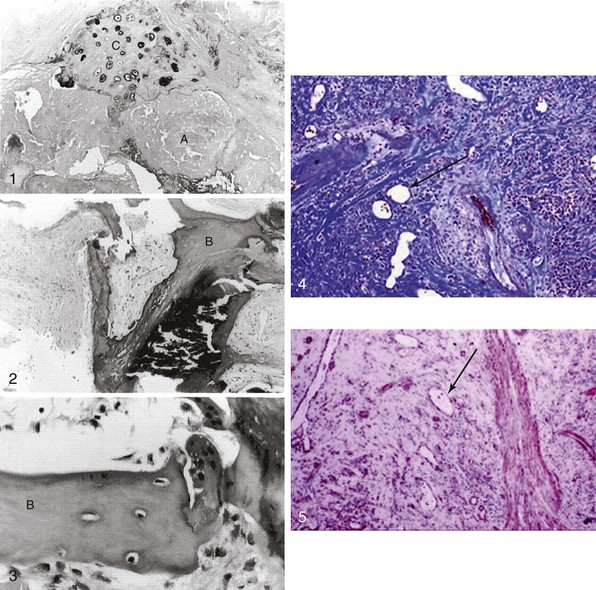
Panel 1 shows atheromatous (A) and chondrocyte-like (C) changes. In contrast, 2 and 3 depict mature bone-like structures (B). 4 and 5 depict massive valvular collagen accumulation/fibrosis (Masson’s trichrome stain) and α-smooth muscle actin (immunohistochemistry) as well as areas of neovascularization (arrows). (Modified from Mohler 3rd ER, Gannon F, Reynolds C, et al. Bone formation and inflammation in cardiac valves. Circulation 2001;103:1522–8; and Rajamannan NM, Nealis TB, Subramaniam M, et al. Calcified rheumatic valve neoangiogenesis is associated with vascular endothelial growth factor expression and osteoblast-like bone formation. Circulation 2005;111:3296–301.)
Experimental Models
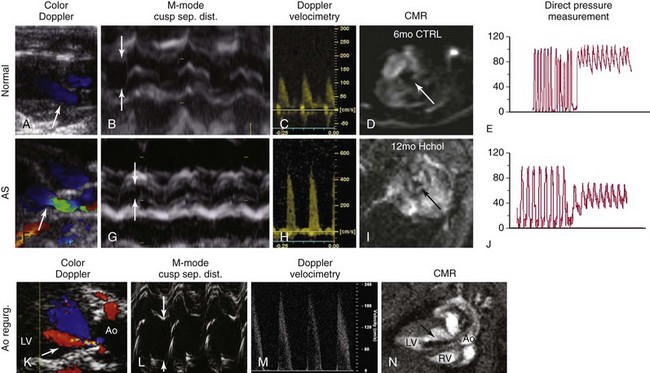
A through C, Measurements of aortic valve function using echocardiography in nonhypercholesterolemic mice (i.e., “normal” animals). Note that there is no evidence of aortic valve regurgitation on color-flow Doppler imaging, cusp separation distance is relatively large (>0.8 mm; arrows), and peak transvalvular velocities are relatively low (<2 m/sec). These data correspond to relatively large aortic valve orifice areas measured on cardiac magnetic resonance (CMR) (D), and low transvalvular pressure gradients measured directly with a Millar catheter (E). F through H, In mice with aortic stenosis (AS), note that there is again no evidence of aortic valve regurgitation (i.e., normal flow in green on the ventricular side of the valve during diastole in F), cusp separation distance is markedly reduced (<0.6 mm; arrows), and peak transvalvular velocity is markedly elevated (>4 m/sec). I, Subsequent measurement of aortic valve area using CMR showed clear reductions in aortic valve orifice area and J, dramatic increases in peak transvalvular pressures. K through N, The importance of using multiple measurements of valve function in mice; the presence of aortic valve regurgitation (Ao regurg) (K, N) dramatically elevates peak transvalvular velocity and gives the appearance of aortic valve dysfunction due to hyperdynamic cardiac function (M). In contrast, aortic valve regurgitation does not influence cusp separation distance (arrows) (L). Ao, Aorta; CTRL, control; Hchol, high cholesterol; LV, left ventricle; RV, right ventricle. (From Miller JD, Weiss RM, Heistad DD. Calcific aortic valve stenosis: methods, models, and mechanisms. Circ Res 2011;108:1392–412.)
Induction of Osteogenic Signaling Cascades
Bone Morphogenetic Protein Signaling
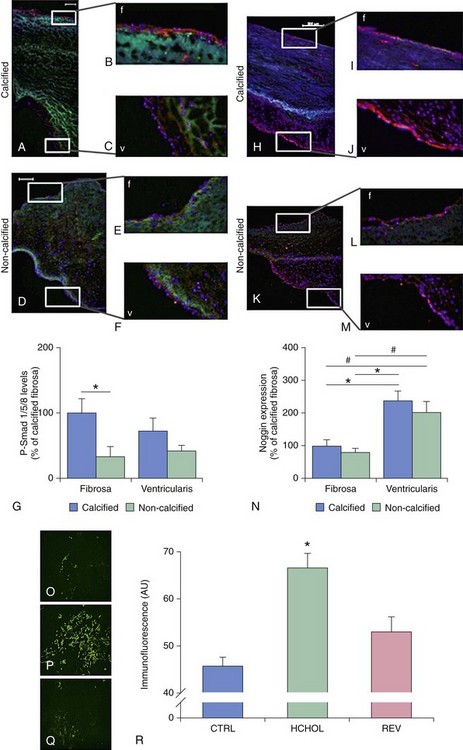
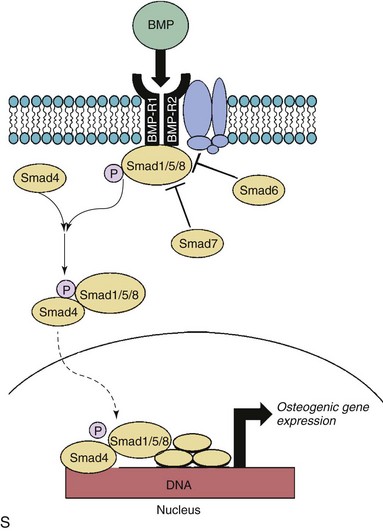
A through G depict changes in levels of phospho-Smad1/5/8 (P-Smad), a key signal transduction molecule in bone morphogenic protein (BMP) signaling, in calcified and noncalcified regions of human aortic valves. H through N depict changes in levels of Noggin (an endogenous inhibitor of BMP signaling) in calcified and noncalcified regions of human aortic valves. Note that P-Smad1/5/8 levels are highest where Noggin levels are lowest, suggesting that reductions in endogenous inhibitors of BMP signaling are a key permissive step for increases in canonical pathway activation. O through R show P-Smad1/5/8 levels in aortic valves from hypercholesterolemic (HCHOL) mice with severe fibrocalcific aortic valve disease. Note that reduction of hyperlipidemia (REV) significantly reduces canonical BMP signaling, suggesting that this pathway is labile even in advanced stages of valve disease. S depicts canonical BMP signaling, in which binding of BMP ligand to its receptor complex results in phosphorylation (P) of Smad1/5/8, translocation of the activated Smad complex to the nucleus, and induction of osteogenic gene expression. (A through N from Ankeny RF, Thourani VH, Weiss D, et al. Preferential activation of SMAD1/5/8 on the fibrosa endothelium in calcified human aortic valves—association with low BMP antagonists and SMAD6. PLoS One 2011;6:e20969; O through R from Miller JD, Weiss RM, Serrano KM, et al. Evidence for active regulation of pro-osteogenic signaling in advanced aortic valve disease. Arterioscler Thromb Vasc Biol 2010;30:2482-24862.)
Wnt/β-Catenin Signaling
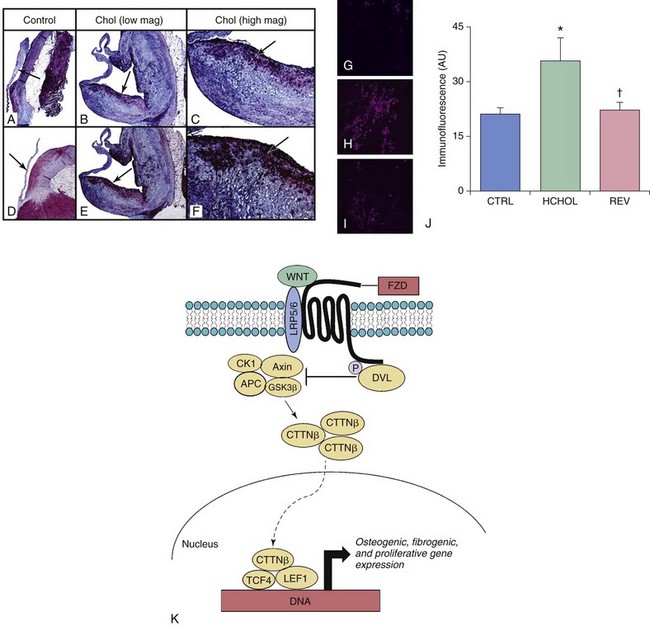
A through F are micrographs depicting changes in Wnt/β-catenin signaling in control rabbits (CTRL) and in rabbits with hypercholesterolemia-induced aortic valve disease (Chol). More specifically, A through C show changes in low-density lipoprotein receptor–related protein 5 (Lrp5), a receptor critical for Wnt ligand binding. D through F show changes in β-catenin levels. Note that levels of both Lrp5 and β-catenin are dramatically increased by hypercholesterolemia and that these increases occur preferentially on the aortic side of the valve. G through J depict changes in β-catenin immunofluorescence in aortic valves from hypercholesterolemic mice. K, Like BMP signaling (shown in Figure 3-5), β-catenin immunofluorescence can be markedly attenuated by a reduction in blood lipids in hypercholesterolemic mice with advanced fibrocalcific aortic valve disease. APC, Adenomatous Polyposis Coli tumor suppressor gene; CK1, Casein Kinase 1; CTTNβ, b-catenin protein; DVL, disheveled proteins; FZD, Frizzled proteins; GSK3β, glycogen synthetase kinase-3β; HCHOL, high-cholesterol diet; LEF1, lymphoid enhancer binding factor; REV, reversed; TCF4, Transcription Factor 4.
Transforming Growth Factor-β Signaling
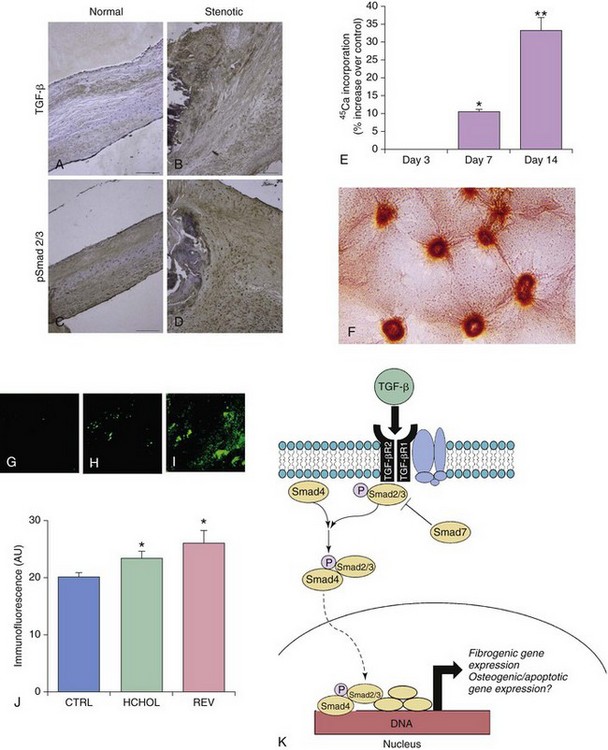
A through D depict changes in TGF-β levels and phospho-Smad2/3 levels in aortic valves from humans with severe fibrocalcific aortic valve disease. Note that phospho-Smad2/3 levels are increased most dramatically in pericalcific regions of the stenotic valve. E and F show changes in calcium ( 45Ca) accumulation (E) and nodule formation (F) in aortic valve interstitial cells treated with exogenous TGF-β1. Note that prolonged exposure to TGF-β1 in cells plated directly on plastic culture plates results in robust calcium accumulation and Alizarin red–positive nodule formation. G through J depict changes in phospho-Smad2 levels in aortic valves from mice with severe fibrocalcific aortic valve disease. Note that canonical TGF-β signaling is increased in valves from mice with severe fibrocalcific aortic valve disease. In contrast to osteogenic signaling, however, reducing blood lipids does not effectively reduce TGF-β signaling in mice with advanced aortic valve dysfunction and stenosis. K depicts canonical TGF-β signaling, in which binding of TGF-β ligand to its receptor complex results in phosphorylation (P) of Smad2/3, translocation of the activated Smad complex to the nucleus, and induction of fibrogenic and osteogenic gene expression. CTRL, Control; HCHOL, high-cholesterol diet; P, phosphorylation; REV, reversed; TGF-βR, TGF-β receptor. (A through D from Osman N, Grande-Allen KJ, Ballinger ML, et al. Smad2-dependent glycosaminoglycan elongation in aortic valve interstitial cells enhances binding of LDL to proteoglycans. Cardiovasc Pathol 2013;22:146–55; E and F from Jian B, Narula N, Li QY, et al. Progression of aortic valve stenosis: TGF-beta1 is present in calcified aortic valve cusps and promotes aortic valve interstitial cell calcification via apoptosis. Ann Thorac Surg 2003;75:457–65; G through J from from Miller JD, Weiss RM, Serrano KM, et al. Evidence for active regulation of pro-osteogenic signaling in advanced aortic valve disease. Arterioscler Thromb Vasc Biol 2010;30:2482–6.)
Modulators of Osteogenic Differentiation
Direct Inhibitors of Osteogenic Signaling
Angiotensin II
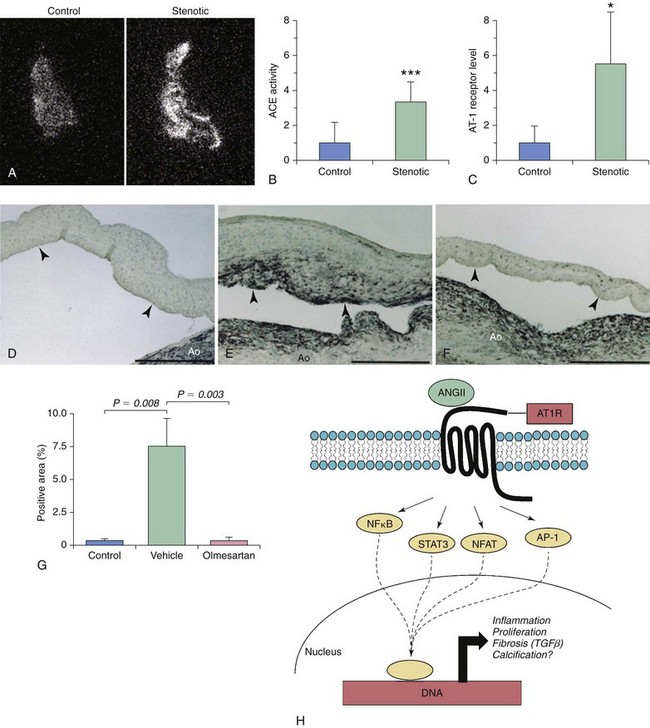
A and B, Autoradiography imaging of angiotensin-converting enzyme (ACE) levels in control and stenotic valves, with quantitative measurement of image intensity in B and C. Note that local ACE levels are significantly increased in stenotic valves and are associated with dramatic increases in angiotensin type 1 receptor (AT1R) levels. D through G illustrate the effects of long-term AT1R inhibition (with olmesartan) on myofibroblast activation in a hypercholesterolemic rabbit model of fibrocalcific aortic valve disease. Note that long-term AT1R blockade significantly reduces the number of α-smooth muscle actin–positive myofibroblasts in this model. H depicts potential signaling cascades that may be activated following binding of ANGII to AT1R. Note that AT1R activation has the potential to elicit a broad range of responses, including induction of cellular inflammation, proliferation, fibrosis, and calcification. AP-1, activator protein-1; NFAT, nuclear factor of activated T-cells; NFκB, nuclear factor κB; STAT, signal transducer and activator of transcription. (A through C from Helske S, Lindstedt KA, Laine M, et al. Induction of local angiotensin II-producing systems in stenotic aortic valves. JACC 2004;44:1859–66; D through G from Arishiro K, Hoshiga M, Negoro N, et al. Angiotensin receptor-1 blocker inhibits atherosclerotic changes and endothelial disruption of the aortic valve in hypercholesterolemic rabbits. JACC 2007;49:1482–9.)
Inflammatory Cell Infiltrate and Proinflammatory Cytokines
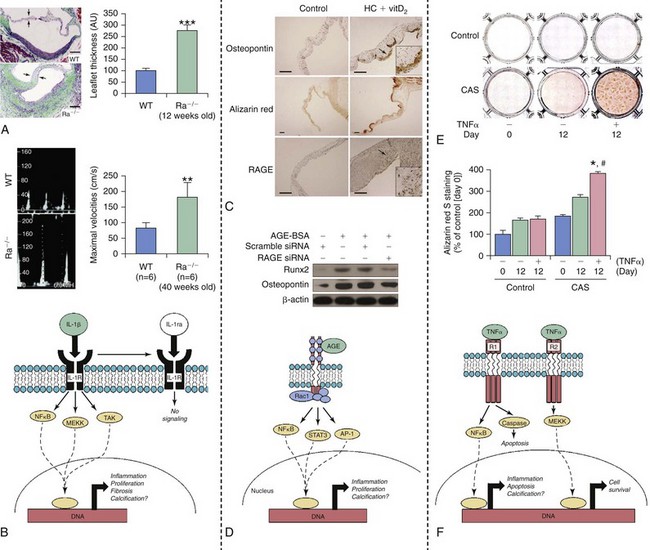
A and B illustrate histopathologic and functional changes in aortic valves from mice that are deficient (Ra−/−) in an endogenous antagonist to interleukin 1 (IL-1ra); WT, wild type mice. Note that increasing IL-1β signaling dramatically increases leaflet thickness (A) and peak transvalvular velocity (B). C and D depict changes in RAGE (receptor for advanced glycosylation end products) signaling in hypercholesterolemic rabbits (HC) receiving high-doses of vitamin D2 (vitD2) to induce fibrocalcific aortic valve disease. Note that both calcium accumulation and induction of osteopontin are associated with increases in RAGE levels in aortic valves from these animals. Furthermore, induction of osteogenic signaling by AGE–bovine serum albumin (AGE-BSA) can be markedly attenuated by knockdown of RAGE in aortic valve interstitial cells in vitro. E and F show changes in tumor necrosis factor α (TNFα)–induced calcification in valve interstitial cells from control/nonstenotic valves and from calcified stenotic valves (CAS). Note that TNFα-induced calcium accumulation is much more dramatic in cells from patients with aortic stenosis, suggesting that genetic and/or epigenetic changes are likely to increase the propensity for valve calcification and osteogenesis even after cells are taken out of the body/”fibrocalcific aortic valve disease milieu” and cultured. AGE, advanced glycosylation end products; AP-1, activator protein-1; MEKK, mitogen activated protein kinase kinase kinase; Rac1, ras-related C3 botulinum toxin substrate 1; Runx2, runt-related transcription factor 2; siRNA, small interfering RNA; STAT, signal transducer and activator of transcription; TAK, transforming growth factor-beta activated kinase. (A and B from Isoda K, Matsuki T, Kondo H, et al. Deficiency of interleukin-1 receptor antagonist induces aortic valve disease in balb/c mice. Arterioscler Thromb Vasc Biol 2010;30:708–15; C and D from Li F, Cai Z, Chen F, et al. Pioglitazone attenuates progression of aortic valve calcification via down-regulating receptor for advanced glycation end products. Basic Res Cardiol 2012;107:306; E and F from Yu Z, Seya K, Daitoku K, et al. Tumor necrosis factor-α accelerates the calcification of human aortic valve interstitial cells obtained from patients with calcific aortic valve stenosis via the BMP2-Dlx5 pathway. J Pharmacol Exp Ther 2011;337:16–23.)![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Cellular and Molecular Basis of Calcific Aortic Valve Disease
 Key Points
Key Points




Only gold members can continue reading. Log In or Register to continue
