Fig. 25.1
Cellular NSIP. The lung shows diffuse involvement of the alveolar interstitium by a mild chronic inflammatory infiltrate
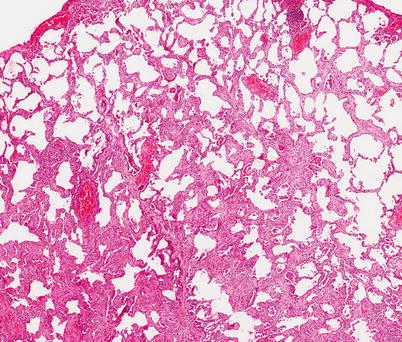
Fig. 25.2
Fibrotic NSIP. The lung shows diffuse involvement of the alveolar interstitium by a homogenous fibrosis with minimal inflammation. No fibroblastic foci were seen at high power (H&E stain ×10 magnification)
High resolution computed tomography (HRCT) is the primary means of detecting SSc-ILD. Interstitial lung disease is present in over 90 % of SSc patients with abnormal pulmonary function tests and in up to 65 % of SSc patients overall [13, 14]. HRCT appearances are typically compatible with NSIP, with prominent ground-glass attenuation and a low prevalence of honeycomb change [15]. HRCT has a high diagnostic sensitivity which has obvious advantages but has also caused difficulties for clinicians as very limited HRCT abnormalities are often disclosed by routine screening tests. As HRCT abnormalities are often limited and difficult to interpret, a multi-disciplinary evaluation of disease severity is essential, with the integration of symptoms, pulmonary function tests and the extent of disease on HRCT. The selection of patients for treatment, discussed later, has been facilitated by studies of the natural history and treated course of SSc-ILD in large historical cohorts [16–18], and especially by recent HRCT data [19, 20].
Risk Factors for SSc-ILD
SSc-ILD has been linked to both the distinction between limited and diffuse cutaneous disease and the autoantibody profile. SSc is sub-classified as limited or diffuse cutaneous disease, according to the extent of skin involvement. In limited disease, skin involvement is distal to the elbows and knees (although facial involvement may occur) whereas in diffuse disease, there is variable involvement of the trunk, shoulders, pelvic girdles and the face and acral areas. In one series, SSc-ILD was identified in 53 % of patients with diffuse disease but in only 35 % of patients with limited disease [21].
It is likely that these observations reflect linkages to autoantibody status. In the EUSTAR database, autoantibody status was a more powerful predictor of major organ involvement than the distinction between diffuse and limited cutaneous disease [21]. Antinuclear antibodies (ANA) are present in more than 90 % of SSc patients. Autoantibodies against topoisomerase I (ATA, also known as anti Scl-70 antibody), present in over 20 % of SSc patients, are associated with the development of pulmonary fibrosis in over 85 % of cases [22]. However, only 40 % of patients with SSc-ILD are ATA positive [23]. Reported correlations between ATA levels and the severity of SSc-ILD [24, 25] are not sufficiently consistent to influence the investigation of individual patients. Anti-centromere antibody (ACA) positivity, associated with a very low prevalence of significant SSc-ILD, is linked to limited cutaneous disease and an increased risk of SSc-PH [4, 26].
Genetic Associations
The accumulated evidence indicative of a genetic predisposition to SSc includes associations between specific autoantibodies and major histocompatibility complex (MHC) classes, clustering of SSc with other rheumatic diseases in family members and familial cases of SSc, including in twins [27]. The Choctaw Indians have a ten-fold increase in prevalence of SSc and a genome-wide screen has disclosed multiple microsatellite markers in chromosome regions associated with SSc, including the MHC, fibrillin 1 gene (15q), the topoisomerase 1 gene (chromosome 20q) and the SPARC gene (secreted protein, acid rich in cysteine; chromosome 5q) [28]. ATA positivity is strongly linked to the carriage of the HLA-DRB1*11 and HLA-DPB*1301 alleles [23]. ATA positivity, diffuse cutaneous disease and SSc-ILD have been associated with the rs763361 single nucleotide polymorphism in the CD226 gene [29]. Other polymorphisms linked to SSc-ILD have included the IL-1α and IL-1β genes [30, 31]. By contrast, single nucleotide polymorphisms in the surfactant protein B gene are associated with a lower prevalence of SSc-ILD in Japanese SSc patients [32].
Clinical Presentation of SSc-ILD
In SSc-ILD, respiratory symptoms are notorious for their non-specificity and variability, with the severity of exercise limitation correlating poorly with disease severity, as judged by pulmonary function tests (PFT) and the extent of disease on HRCT. The difficulty in evaluating symptoms lies in the fact that in SSc, dyspnoea may arise from a multiplicity of causes including interstitial lung disease, pulmonary vascular limitation (even when overt PH is not present), cardiac involvement, musculoskeletal disease, loss of fitness due to general debility and anemia, with more than one factor often coexisting. In some patients, severe systemic disease results in a major reduction in daily activity and loss of pulmonary reserve is not exposed. By contrast, knowledge of the likelihood of lung involvement leads to concerns about exercise intolerance in other cases, even when interstitial lung disease is absent or relatively limited. Accurate assessment of dyspnoea requires observation of the exercising patient. The absence of oxygen desaturation during a 6 min walk test (or a more strenuous form of exercise) suggests that dyspnoea largely results from extra-pulmonic factors.
A detailed history should include occupational exposures known to result in SSc and the impact of lung symptoms on quality of life. The duration of systemic disease (as judged by SSc symptoms and not by Raynaud’s phenomenon) may influence treatment decisions, as discussed later. It is also important to explore the evolution of dyspnoea: a long-term lack of change in exertional dyspnoea is reassuring in patients with severe SSc-ILD. Recently, aspiration due to gastro-esophageal regurgitation (GER) has been recognized as a potential trigger of SSc-ILD. GER may cause troublesome cough (due to a vagally-mediated cough reflux) and in occasional patients, episodes of wakening with a choking sensation may be indicative of significant nocturnal aspiration of gastric contents.
Physical examination does not contribute greatly to the assessment of SSc-ILD. The classical finding of fine bi-basal crackles is often absent in limited SSc-ILD. Changes in the intensity of crackles have not been validated as a reliable indicator of disease progression. Finger clubbing is very rare in SSc-ILD. In end-stage SSc-PH, positive clinical findings include a loud pulmonary component of the second heart sound, a right ventricular heave, elevated jugular venous pressure, signs of peripheral oedema – but these signs are not reliably present in less advanced PH. Impairment in chest wall movement due to severe thoracic skin involvement is a rare extra-pulmonic cause of exertional dyspnea.
Pulmonary Function Tests (PFTs)
PFTs have been used historically for both the staging of disease severity and the serial monitoring of SSc-ILD. It is generally accepted that in these regards, PFT are more reliable than symptoms or chest radiography. However, the limitations of PFT need to be appreciated by the clinician. In staging severity, the normal PFT range, varying from 80 to 120 % of expected values based on age, height and gender [33], is a major confounder. For example, an FVC value of 75 % of predicted may equally represent a relatively minor fall of 5 % or a very major reduction of 45 % from premorbid values of 80 and 120 % respectively. Thus, it is essential that the evaluation of disease severity should be a multidisciplinary exercise, with the integration of PFT, HRCT findings and symptoms. However, it can at least be concluded that severe reductions in lung volumes and measures of gas transfer are reliably indicative of severe pulmonary disease.
The classical PFT profile in SSc-ILD is a restrictive ventilatory defect, with reduced total lung capacity, reduced forced vital capacity (FVC), an FEV1/FVC ratio of >0.8, reduced carbon monoxide diffusing capacity (DLco) and reduced lung compliance. Moderate restriction (FVC 50–75 % of predicted) is found in up to 25–30 % of SSc patients, with 10–15 % having severe restriction [4]. DLco estimation and can be viewed as a “gestalt” evaluation of resting pulmonary function, as it captures both ventilatory defects and reductions in blood volume within ventilated lung. Disproportionate reductions in DLco (when compared to lung volumes) can arise in two distinct scenarios. In smokers, the coexistence of interstitial lung disease and emphysema (widely known as the “combined pulmonary fibrosis and emphysema syndrome”) results in preservation of lung volumes (even when both processes are extensive) but a devastating reduction in DLco, a combination best documented in idiopathic interstitial pneumonia [34] but also seen in SSc-ILD [35]. A more frequent scenario in SSc-ILD (and in SSc in general) is disproportionate reduction in DLco due to significant pulmonary vascular limitation (with or without overt SSc-PH). In recent series, elevation of the FVC/DLco ratio has been used as a marker of pulmonary vascular limitation [36, 37]. However, there are theoretical advantages in an alternative variable, the gas transfer coefficient (Kco), which quantifies carbon monoxide uptake per unit volume of ventilated lung. It is often overlooked that DLco is calculated as the product of measured Kco and measured VA [38] (accounting for the higher measurement variability of DLco than other pulmonary function variables). Thus, the use of the FVC/DLco ratio depends upon the accurate measurement of three variables (Kco, VA and FVC) whereas Kco carries the measurement variation of only one manoeuvre.
Spirometric volumes are highly reproducible in laboratories with an acceptable level of quality assurance. Body plethysmography is a more complex measurement performed inside a sealed, air-tight chamber and is used to estimate total lung capacity (TLC) and residual volume (RV). In interstitial lung disease, reductions in TLC and RV tend to mirror reductions in FVC and in most cases add little to FVC measurement. However, plethysmography should be performed at presentation in order to allow an alternative monitoring variable to be used in occasional patients, in whom forced spirometric manoeuvres are contraindicated by glaucoma, significant chest wall discomfort or severe microstomia.
In SSc-ILD, resting arterial gases tend to be normal in mild to moderate disease except when there is concurrent pulmonary hypertension. In advanced disease, hypoxia is usually associated with hypocapnoea (reflecting alveolar hyperventilation). The performance of arterial gases can generally be avoided in routine evaluation as simple oximetry is an adequate substitute, although sometimes confounded by Raynaud’s phenomenon. Ear lobe capillary gases, which can be measured in many lung function laboratories, are also an acceptable substitute for arterial gases.
Maximal exercise testing adds little to the routine evaluation of SSc-ILD. However, in occasional patients with exertional dyspnea that is disproportionate to the severity of SSc-ILD, maximal exercise testing is a useful means of excluding clinically significant interstitial lung disease. The absence of oxygen desaturation or widening of the alveolar-arterial oxygen gradient at end exercise on room air may allow the clinician to conclude that exercise tolerance is limited by extra-pulmonary factors such as musculoskeletal disease or lack of fitness. The six minute walk test is more useful as it more closely approximates daily activity. Major desaturation should prompt the clinician to exclude SSC-PH and to consider the potential benefits of ambulatory oxygen.
In routine monitoring, serial pulmonary function tests have a central role. The normal range is no longer a major confounder as significant change is indicative of disease progression, irrespective of premorbid pulmonary function levels. However, as in interstitial lung disease in general, measurement variation creates difficulties. Serial PFT trends are reliably indicative of disease progression only when FVC change exceeds 10 % of baseline values (e.g. a change from 2.0 to 1.8 l). DLco trends may also be helpful but are less specific when there is concurrent pulmonary vascular limitation. Even when pulmonary function trends are significant, it is important that alternative explanations for functional decline are considered, including infection, pulmonary embolism and cardiac disease. It is important to remember that measurement variation can result equally in the under-statement of change. Lesser changes (e.g. a 5–10 % change in FVC) may be indicative of disease progression. Thus, functional trends should be reconciled with symptomatic change and, in selected cases, serial imaging data.
Imaging
High resolution computed tomography (HRCT) can now be viewed as the reference standard for the detection of SSc-ILD. The chest radiograph is insensitive in the detection of SSc-ILD [39] although useful as a screening tool. HRCT findings closely resemble those seen in idiopathic NSIP [15] typically consisting of a variable mixture of ground-glass attenuation and reticulation (Fig. 25.2). In a minority of patients with overt honeycomb change, a histological pattern of usual interstitial pneumonia can be suspected. The historical belief that ground-glass attenuation is indicative of reversible inflammatory disease has not stood the test of time. In occasional patients with prominent ground-glass, without associated reticulation or traction bronchiectasis, disease is, indeed, likely to be reversible (Fig. 25.3a). However, in the great majority of cases, ground glass is admixed with reticulation and there is traction bronchiectasis (Fig. 25.3b), a combination of HRCT signs that is reliably indicative of fine fibrosis [13, 40, 41].
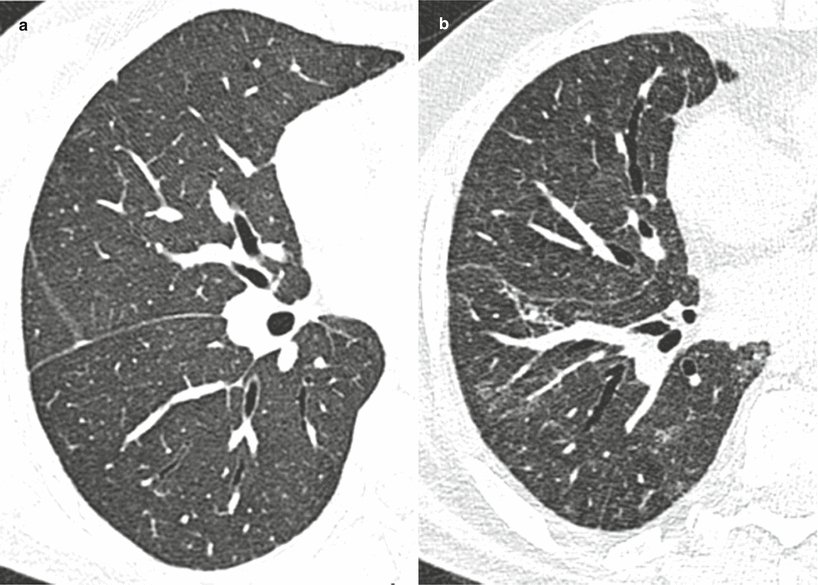
Fig. 25.3
(a) HRCT appearances in a patient with biopsy-proven cellular NSIP. There is a diffuse increase in lung attenuation without admixed reticulartion or traction bronchiectasis. (b) HRCT appearances in a patient with biopsy proven fibrotic NSIP. The diffuse increase in attenuation on HRCT represents fine fibrosis, with the presence of obvious traction bronchiectasis an important clue that interstitial disease was likely to be irreversible
Apart from the detection of disease, HRCT provides an alternative means of evaluating disease severity. Precise quantification of disease extent is arduous and insufficiently “user-friendly” to be a part of routine evaluation. However, rapid semi-quantitative assessment of disease extent on HRCT helps the clinician to address the confounding effect of the normal range in the interpretation of pulmonary function tests. As discussed later, HRCT can also be used to stage disease as mild or extensive.
Serial HRCT evaluation tends to be over-used by clinicians, based on the supposition that a sensitive test must add to the accuracy of monitoring. In reality, HRCT is often too sensitive in the detection of change. No definition of “significant” HRCT change has been validated and therefore subtle regional HRCT change in patients with stable pulmonary function tests is difficult to interpret. In other patients with major pulmonary function trends, there may be little or no change on HRCT. Furthermore, the long term risk of malignancy with excessive exposure to radiation should not be overlooked. Thus, the inclusion of HRCT in a routine monitoring protocol is difficult to justify. Serial HRCT should only be performed on a case by case basis to answer specific clinical questions, with the most frequent scenarios being discordance between symptomatic change and pulmonary function trends and disproportionate decline in measures of gas transfer, ascribable equally to progression of interstitial lung disease and worsening of pulmonary vascular disease.
Prognostic Evaluation of SSc-ILD: When Should Treatment Be Instituted?
The routine use of HRCT in the initial evaluation of SSc often discloses limited interstitial abnormalities of uncertain significance. This creates a major dilemma for the clinician. It is axiomatic that early treatment is needed when disease is intrinsically progressive. However, when abnormalities are mild or “sub-clinical”, overly aggressive intervention can result in major side-effects without therapeutic gain. Intrinsically progressive disease cannot be identified reliably. However, based on accumulated clinical experience, the decision to institute therapy should be influenced by the severity of lung disease, the duration of systemic disease and evidence of ongoing disease progression.
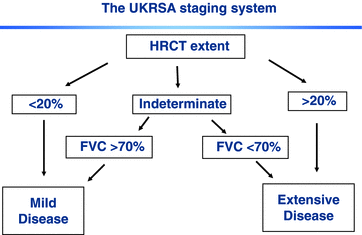
It is widely accepted that the threshold for treatment is critically dependent on disease severity. Severe disease is a marker of repeated past disease progression and is, therefore, indicative of an increased likelihood of future disease progression. Furthermore, in severe disease, further progression is associated with major changes in exercise tolerance and quality of life. In a staging system centred on disease severity, Goh and co-workers evaluated the prognostic value of candidate FVC and HRCT disease extent thresholds [20]. Key prognostic thresholds consisted of a percent predicted FVC value of 70 % and an HRCT extent threshold of 20 % (i.e. 20 % of the total lung volume). In the staging system for SSc-ILD shown in Box 25.1, lung disease was classified as “mild” or “extensive”, based on rapid semi-quantitative HRCT evaluation and in cases with an “indeterminate” disease extent, an FVC threshold of 70 %. These thresholds were very similar to HRCT and FVC thresholds in the Scleroderma Lung Study (SLS), below which treatment effects with oral cyclophosphamide were seen [19]. The two studies establish that the staging of severity using HRCT and FVC data is likely to be useful in informing treatment decisions in clinical practice. However, it should also be stressed that despite its prognostic utility, the Goh staging system has yet to be integrated into a validated management algorithm.
Box 25.1
The mild/extensive severity staging system for prognostic evaluation in SSc-ILD. Extensive disease is associated with an increase in mortality of over three-fold and a much higher likelihood of disease progression in the next year.
The duration of systemic disease is also an important consideration as the risk of progression of SSc-ILD is greater early in the course of systemic disease. Steen and colleagues observed that the risk of progression is highest in the first 4 years of systemic disease and especially in the first 2 years. The risk is even greater when the onset of lung disease precedes the cutaneous manifestations of SSc [4]. With regard to treatment decisions, interstitial lung disease that is detected early in the course of systemic disease can be viewed as intrinsically progressive, with a reduced threshold for introducing therapy. By contrast, in patients with minor pulmonary function impairment after more than 5 years of systemic disease, mild SSc-ILD is less likely to evolve to severe fibrotic lung disease.
Lastly, recent progression of disease, as judged by a variable combination of serial PFT tends, serial imaging data and symptomatic change, is, in itself, an indication for therapy. In SSc-ILD, the exact value of recent disease progression as a malignant prognostic determinant has yet to be quantified in clinical series. However, recent disease progression has been predictive of increased mortality in other forms of interstitial lung disease (most widely studied in idiopathic pulmonary fibrosis) [42]. Intervening to stabilize disease that is overtly progressive is warranted on simple commonsensical grounds.
Thus, the severity of disease, the duration of systemic disease and evidence of recent progression should all be taken into account when treatment decisions are made. Currently, no validated algorithm exists to incorporate all these factors into management. Treatment decisions must be made on a case by case basis, acknowledging the wishes of the patient (which often become the key determinant when the grounds for introducing therapy are marginal). When immediate treatment is not warranted, rigorous monitoring is essential, primarily based on the performance of serial PFT, with an intention to treat if disease progression becomes evident. Based on accumulated clinical experience, both in SSc-ILD and idiopathic interstitial lung disease, 3–6 monthly repetition of PFT is recommended with the time interval between PFT prolonged after disease has been stable for at least 2 years.
As in interstitial lung disease in general, a biomarker in SSc-ILD that accurately predicted disease progression would greatly increase the accuracy of treatment decisions. At present, no such biomarker is exists. Rapid clearance of inhaled technetium-labeled diethylene-triamine-pentacetate (99mTc-DTPA) from the lungs (a marker of increased alveolar cell permeability) is associated with a shorter time to decline in FVC in SSc-ILD, before and after adjustment for disease severity [43]. As this test is not widely available and the associated radiation burden is significant, substitute serum biomarkers are needed to detect lung epithelial damage. Attention has focused on two lung glycoproteins, KL-6 and SP-D in SSc-ILD [44–46] but the utility of these and other candidate biomarkers in routine practice is uncertain. With regard to non-epithelial biomarkers, IL6 was evaluated in a large retrospective study of SSc-ILD patients, including over 200 patients in a separate “test cohort”. Increased serum IL6 levels were associated with more rapid disease progression, especially in mild disease [47].
Bronchoalveolar lavage (BAL) cellularity has been viewed historically as an invaluable aide to treatment decisions in patients with SSc-ILD. In a number of small cohorts, a BAL neutrophilia was linked to a worse outcome. However, these observations took no account of the now well-recognised association between the presence of a BAL neutrophilia and extensive SSc-ILD. Based on findings in two large patient cohorts, it appears that the link between disease progression and a BAL neutrophilia merely reflects the fact that more extensive SSc-ILD is more intrinsically progressive. In over 140 patients with SSc-ILD, BAL neutrophil levels were linked to global disease severity, as judged by PFTs and HRCT disease extent, and had no independent prognostic value with regard to disease progression or long term mortality [48]. In the SLS, PFT follow-up was such shorter short in duration but was carried out at strictly standardized time intervals. BAL findings were predictive neither of a treatment effect nor of disease progression in the placebo arm [49]. Following these studies, BAL is now performed much less frequently in the prognostic evaluation of SSc-ILD. BAL continues to be performed in selected patients with disproportionate upper lobe abnormalities (to exclude pulmonary tuberculosis) or when HRCT evaluation suggests a coexisting disease process such as smoking related interstitial lung disease.
Management
Historically, the core principle in the management of SSc-ILD has been to suppress inflammation with corticosteroid or immunosuppressive therapy. This approach, based on a disease model in which inflammation precedes and leads to fibrosis, has been supported only by anecdotal reports and uncontrolled treatment effects in small groups of patients. The key limitation is the low prevalence of SSc-ILD in routine practice. For this reason, treatment statements were determined by clinical experience at single referral centres for many years. However, since the millenium, multi-centre treatment studies in SSc-ILD have proven to be possible. Placebo-controlled trials of oral cyclophosphamide [19], intravenous cyclophosphamide [50] and bosentan [51] have now been completed.
Cyclophosphamide was a logical trial therapy in the first controlled treatment trials in SSc-ILD because partial regression with treatment was seen in some patients in small pilot series. In the landmark placebo-controlled SLS trial of oral cyclophosphamide, statistically significant treatment effects were apparent at 1 year on FVC levels, dyspnoea, skin thickening and quality of life [19]. The SLS was followed by a UK placebo-controlled trial of intravenous cyclophosphamide (given once a month for 6 months, followed by maintenance therapy with oral azathioprine) [50]. The study was under-powered due to recruitment difficulties that are now regarded as inescapable in this field. The FVC treatment effect was similar to that seen in the SLS trial, although only marginally significant (p = 0.08) due to the small cohort sizes (n = 45) [50]. Taken together, the two studies prompted EULAR to conclude that cyclophosphamide was an appropriate therapy in SSc-ILD [52].
However, this conclusion has not been uniformly accepted and at the least, it is clear that cyclophosphamide should not be introduced indiscriminately in all patients with SSc-ILD. In both trials, the average FVC treatment benefit was less than 5 % of baseline values and in the SLS trial, although not in the UK trial, the small gain in FVC came at the cost of a significant prevalence of adverse effects. Importantly, many patients with mild lung disease were enrolled in both studies. This is understandable: the risk that an individual patient may receive a placebo, when open therapy is available, is likely to be more acceptable to patients and referring physicians alike when lung disease was not overtly progressive or severe. In keeping with this limitation, it is salutary that after patients in the SLS trial had completed treatment and returned to routine follow-up, less than 15 % were prescribed open therapy by their primary physicians [53]. Crucially, there was no treatment effect in the SLS trial in patients with mild disease on HRCT. By contrast, there was a striking treatment effect on FVC (>10 %) in extensive fibrotic disease, providing a useful clue as to which patients are likely to benefit in clinical practice [19].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


