CHAPTER 122 Interrupted Aortic Arch
HISTORICAL NOTES
The first description of an interrupted aortic arch is attributed to Steidele in 1778.1 That case involved absence of the aortic isthmus. Absence of the aortic arch segment between the left common carotid artery and the left subclavian artery was described by Seidel in 1818.2 Absence of the aortic segment between the innominate artery and the left common carotid artery was described by Weisman and Kesten in 1948.3 In 1955, Samson and colleagues performed the first successful surgical repair of interrupted aortic arch in a 3-year-old child.4 The anatomy included absence of the aortic isthmus, the presence of a patent ductus arteriosus, and two ventricular septal defects (VSDs). In the initial operation, the ductus was divided, and then it was joined to the underside of the proximal left subclavian artery, creating luminal continuity between the ascending aorta and the descending aorta. The VSDs were closed 4 years later. Case reports in the 1960s described interposition of prosthetic grafts to bridge the gap between aortic segments. The first use of turned-down aortic arch branches (left subclavian or left common carotid artery) to achieve end-to-end anatomosis to the descending thoracic aorta was described by Sirak and coworkers in 1968.5 One of Sirak’s three patients was the first neonate to survive operation. In 1970, Litwin and associates palliated a neonate with arch interruption and VSD by interposition of a prosthetic tube graft between the proximal main pulmonary artery and the descending thoracic aorta, with banding of the distal main pulmonary artery.6
In 1970, Barratt-Boyes and colleagues7 described successful repair of interrupted aortic arch and simultaneous correction of intracardiac lesions (VSD and total anomalous pulmonary venous connection). Through a left thoracotomy incision, a 12-mm prosthetic tube graft was connected to the descending thoracic aorta. Then, through a median sternotomy, the proximal end of the conduit was connected to the ascending aorta. Hypothermic circulatory arrest facilitated this anastomosis and the repair of the intracardiac lesions. Trusler and Izukawa, in 1975,8 were the first to accomplish direct anastomosis of the descending aorta to the ascending aorta and tranverse arch (with excision of all ductal tissue and with no interposition graft), together with VSD closure. The operation was accomplished through a median sternotomy approach, using cardiopulmonary bypass and deep hypothermic circulatory arrest. Primary repair via median sternotomy using continuous cardiopulmonary bypass (without a period of circulatory arrest) was reported by Asou and colleagues in 1996.9
In the mid 1960s, Angelo DiGeorge, a pediatric endocrinologist, described the association of hypoparathyroidism (with hypocalcemia), thymic aplasia, cleft lip and palate, and altered immunity (eventually recognized to be the consequence of a T-cell abnormality). Several of the patients with this constellation had cardiac anomalies—the most prevalent among DiGeorge’s patients was interrupted aortic arch. DiGeorge syndrome and the closely related velocardiofacial syndrome were subsequently found, in the majority of instances, to be related to a chromosomal deletion in the region of 22q11.2.10–13
ANATOMY AND NOMENCLATURE
The definition of interrupted aortic arch adopted in 2000 by the International Congenital Heart Surgery Nomenclature and Database Project is “loss of luminal continuity between the ascending and descending aorta.”14 Absence of luminal continuity would also be an acceptable description. As a congenital defect, this represents a developmental abnormality of the embryologic arch elements. In the instance of left-sided aortic arch, the proximal arch (between the normally positioned innominate artery and the left common carotid artery) is derived from the aortic sac. The distal arch (between the left common carotid artery and the left subclavian artery) is derived from the left fourth embryonic arch, and the isthmus (between the left subclavian artery and the descending thoracic aorta) from the junction of the left sixth embryonic arch (ductus arteriosus) with the left dorsal aorta and the left fourth embryonic arch. It is therefore not surprising that many anatomic variants of IAA are observed, with respect to both the site of discontinuity and the sites of origin of brachiocephalic vessels.
Aberrant origin of the right subclavian artery from the descending thoracic aorta is encountered frequently in some forms of IAA, and very rarely in others.15 It is generally associated with hypoplasia or obstruction of the left ventricular outflow tract (LVOT), as there is less antegrade flow from the left ventricle. Rarely, the site of origin of the right subclavian artery is a persistent right-sided ductus. Interrupted arch may occur with a right aortic arch, with persistence of a right-sided ductus (or bilateral ducts).
In nearly 100% of cases, loss or absence of luminal continuity reflects complete absence of one element of the aortic arch. In very rare instances, a fibrous strand connects the ascending aorta and associated arch elements with the descending aorta. By convention, these rare cases may be referred to as interrupted aortic arch, although some would make a distinction and refer to such cases as atresia of a particular portion of the arch. Celoria and Patton originally described the most widely accepted classification for IAA in 1959.16 They divided interrupted arch anomalies into three varieties, based on the site of aortic arch interruption (Fig. 122–1). In the common circumstance of left-sided aortic arch, type A refers to interruption that is distal to the left subclavian artery. In type B, the interruption is between the left common carotid and the left subclavian artery. Finally, in type C, the interruption is between the innominate and left carotid arteries. In a multi-institutional outcome study of 472 patients with IAA by the Congenital Heart Surgeons’ Society (CHSS), McCrindle and associates reported that type A was observed in 28% of patients, type B was by far the most common in 70%, and type C the rarest, seen in only 1%.17 An aberrant right subclavian artery, originating from the descending aorta and taking a retroesophageal course, is frequently seen in patients with IAA, particularly type B.15 In another variation, the aberrant right subclavian artery can originate from the right pulmonary artery via a persistent right-sided patent ductus arteriosus; this variation is often called isolated subclavian artery. These variations have been reported and analyzed, and attempts have been made to expand on the classification system of Celoria to include a description of the origin of the right subclavian artery. The classification system reported by Dische and coworkers in 197518 added the subscript 2 to the letter A, B, or C from the Celoria classification when the aberrant right subclavian artery originated from the descending aorta distal to the interruption. Thus, in the system described by Dische, in type A2, the interruption is distal to the left subclavian artery, and the only major arch vessel taking origin from the aorta distal to the interruption is the aberrant right subclavian artery. Similarly, in type B2, both carotid arteries arise from the aortic arch proximal to the interruption and both subclavian arteries arise from the aorta distal to the interruption. In 1982, the nomenclature system of Oppenheimer-Dekker and colleagues19 offered an alternative subclassification also based on the system of Celoria and Patton. This system also includes subdivisions based on the presence of an aberrant right subclavian artery, but here nine types of IAA are classified. Types A, B, and C are determined based on the system of Celoria. Then, no subscript is used if the right subclavian artery has a normal origin, subscript-1 is used when the aberrant right subclavian artery originates from the descending aorta, and subscript-2 is used when the aberrant right subclavian artery arises from the right pulmonary artery via ductal tissue. Most common usage involves the A,B,C system of Celoria together with a specific verbal description of abnormalities of the origin of brachiocephalic vessels, branch pulmonary arteries, or patent ductus arteriosus.
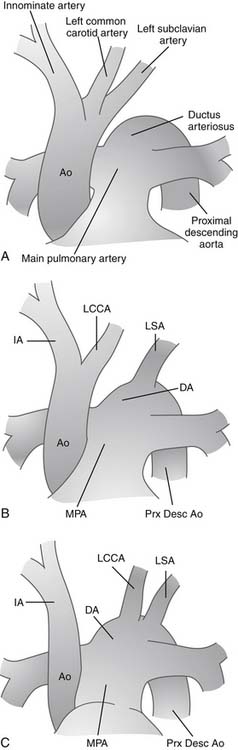
The following is the classification of interrupted aortic arch used in the Diagnostic Long List of the version of the International Paediatric and Congenital Cardiac Code (IPCCC) derived from the nomenclature of the International Congenital Heart Surgery Nomenclature and Database Project of the European Association for Cardio-Thoracic Surgery and the Society of Thoracic Surgeons20:
Interrupted aortic arch complexes nearly always have a large VSD (the exception being cases of IAA with aortopulmonary window, of which a majority have an intact ventricular septum). In the setting of IAA with VSD, the VSD may be of any type. Most commonly, it is of the conoventricular type. Frequently, the conal (outlet) septum is malaligned posteriorly and leftward with respect to the true trabecular septum.21,22 The malalignment of the conal septum is associated with varying degrees of subaortic obstruction.23–25 When a malalignment-type VSD is present, the conal septum not only may be misplaced but also may be hypoplastic. In an echocardiographic study of 53 patients with interrupted aortic arch with VSD, Chin and Jacobs found that 43 of 45 patients with type B IAA had VSDs involving maldevelopment of the outflow region. In type A IAA, only four of eight had this type of VSD.26 In the complex of the left side of the heart and the aorta (the left heart–aorta complex), other levels of obstruction that are often associated with IAA include a prominent and obstructive anterolateral muscle of the left ventricle (muscle of Moulaert), aortic valvar stenosis with or without aortic annular hypoplasia, and fibrous or fibromuscular subaortic stenosis.27 Interrupted aortic arch can coexist with a wide variety of cardiac lesions, as evidenced by the first report from the CHSS in 1994. Jonas and associates15 reported that among 250 cases of IAA at participating centers, associated cardiac anomalies included VSD (183 [73%]), truncus arteriosus (25 [10%]), aortopulmonary window (10 [4%]), univentricular atrioventricular connection (9 [4%]), transposition of the great arteries with VSD (8 [3%]), double-outlet right ventricle (5 [2%]), Taussig-Bing anomaly (4 [2%]), complete atrioventricular canal defect (1 [0.4%]), and congenitally corrected transposition of the great arteries (1 [0.4%]). Isolated IAA (without VSD or associated anomalies) was present in only 4 of 250 patients [2%]). More unusual associations have been the subject of individual case reports, including several cases of aortic atresia with IAA and VSD, in which flow to the ascending aorta and thus the coronary arteries depended on flow either through the circle of Willis or through a patent right-sided ductus arteriosus.
PRESENTATION AND PREOPERATIVE MANAGEMENT
Despite the markedly abnormal circulatory arrangement, manifestations of the abnormal physiology may be subtle in the early neonatal period. If ductal patency persists, typical signs of congestive heart failure may emerge in the first few weeks of life, as the pulmonary vascular resistance begins to fall and pulmonary blood flow increases. More typically, and importantly, constriction of the ductus arteriosus may begin and progress any time in the first days or weeks of life. This may be manifest as a mottled or gray appearance of the lower body, and it is accompanied by signs of congestive heart failure (tachypnea, tachycardia, hepatomegaly, and poor feeding) and then of circulatory insufficiency (irritability progressing to lethargy, oliguria, and then metabolic acidosis and profound shock with multiple organ dysfunction and coagulopathy). Early visual evidence of the difference in oxygen saturation between the upper and lower body may be present. Neither this nor differential blood pressure between upper and lower extremities is a constant or particularly useful finding, as either may be influenced by the dynamic variability of the Qp/Qs, by diminished cardiac performance, and by abnormal anatomy, including aberrant origin of the right subclavian artery (which, in the setting of IAA type B or type C, results in all four extremities having postductal blood pressures). In the absence of a prenatal diagnosis, about half of patients present during the first day of life, and nearly all do within the first 2 weeks of life. In rare instances, persistent patency of the ductus arteriosus is associated with a later presentation.
Echocardiography with color flow mapping is the primary diagnostic study in virtually all cases. When the diagnosis is suspected or established, evaluation as an inpatient in an intensive care setting is advised. Intravenous prostaglandin E1 should be administered promptly to maintain patency of the ductus arteriosus.28 The need for an arterial line and assisted ventilation can be judged best from the initial arterial blood gas measurement. Unless a period of shock has accompanied ductal constriction, pharmacologic support with catecholamine infusions is rarely indicated. In recent years, the use of milrinone, a phosphodiesterase-3 inhibitor, as a continuous infusion has achieved wide acceptance. It probably contributes to the improvement of myocardial performance that generally occurs with correction of acidosis, and it has the added effect of reducing systemic vascular resistance. Assessment of the presence and degree of secondary organ dysfunction includes laboratory evaluation of metabolic status, serum indicators of renal and hepatic function, and a coagulation profile. Hypocalcemia, a frequent finding, may indicate the presence of DiGeorge syndrome, including the hypoparathyroidism phenotype. Fluorescent in situ hybridization (FISH) can reveal the typical hemizygous 22q11.2 deletion seen in 85% to 95% of patients with DiGeorge syndrome.10
In most cases, the key features of the anatomy are revealed by a detailed echocardiographic evaluation. A complete study should clarify the site of aortic interruption and the sites of origin of the arch branches. Additional essential information includes the presence and nature of atrial and ventricular septal defects, and a detailed assessment of the LVOT. This includes measurement of the dimensions of the subaortic region (including a description of the outlet or conal septum, which may be either thickened or hypoplastic, and which may be posteriorly malaligned, thus narrowing the caliber of the subaortic LVOT), as well as the aortic valve anulus and orifice, and the sinotubular junction and ascending aorta.29 An assessment is made of the mitral valve, including the size of its orifice, its competence, and the nature of the subvalvar apparatus. Size of the left ventricle is measured, and a determination is made of whether it extends to the apex of the ventricular mass. Important associated diagnoses to consider include truncus arteriosus communis,30 aortopulmonary window,31,32 transposition of the great arteries, and various forms of univentricular atrioventricular connection. A general visual or quantitative assessment of myocardial contractility is made. Although color flow mapping with Doppler studies can be helpful in evaluating some intracardiac obstructive lesions, the definitive echo assessment is most often made in the setting of a widely patent ductus (and, in most cases, a nonrestrictive interventricular communication). As a result, quantitation of the degree of LVOT obstruction by estimation of a gradient in that region can be misleading.
In some cases, cardiac catheterization with angiography provides important additional information. In particular, it may be helpful in clarifying instances of discontinuous branch pulmonary arteries, anomalous pulmonary venous connections, or transposition of the great arteries (in which case, balloon atrial septotomy may be beneficial). In cases with coexistent aortic atresia, it may help to define the source of coronary blood flow. Under very unusual circumstances, temporary stenting of the ductus may be indicated. (One of us used this strategy in a case of very late presentation of an infant with IAA type A with VSD, who presented with a restrictive duct and respiratory syncytial virus pneumonia. Ductal dilation and stenting provided temporary palliation, making it possible for arch repair and VSD closure to be performed weeks later.) Increasingly, both computed tomography (CT) and magnetic resonance imaging (MRI) are being used to further clarify the anatomy of complex cardiac anomalies in neonates and infants.33 Each technology lends itself to three-dimensional reconstruction, which can be very helpful in clarifying anatomic details, with particular attention to spatial relationships. CT has the advantage of rapid data acquisition, and in some instances it can be accomplished without general anesthesia. It has the disadvantage of radiation exposure. MRI does not involve radiation exposure, but it generally requires the patient to be anesthetized for the study.
SURGICAL MANAGEMENT
Indications and Timing of Surgery
Interrupted aortic arch is incompatible with life without patency of the ductus arteriosus or an alternative pathway for perfusion of the lower body. Before the availability of prostaglandins, the diagnosis of IAA was a surgical emergency. Most often, neonates presented in very poor condition, with a closing ductus, and resuscitative measures had very little efficacy. Patients had to be taken to surgery in very poor condition. Emergent palliative operations were performed under less than ideal conditions and often yielded less than optimal outcomes. In the current era, restoration of ductal patency after infusion of prostaglandin E1 is generally accomplished, facilitating resuscitation in an intensive care unit.28 Recovery of renal and hepatic function usually follows correction of acidosis and optimization of respiratory parameters and fluid and metabolic status. This then facilitates stabilization and assessment of neurologic status, and recovery or correction of coagulation disorders. Genetic evaluation can be initiated, and meaningful family education and counseling can take place before surgery is undertaken. An operation is usually indicated as soon as these preliminary objectives have been achieved, as there is no definitive medical therapy for IAA.
Operative Management
As noted, the history of surgery for IAA began during an era when the use of cardiopulmonary bypass to accomplish repair of intracardiac defects in infants and neonates was a theoretical goal for the future rather than a therapeutic reality. As a result, most early arch repairs were closed-heart (i.e., non-bypass) procedures.6,34 Arch repair was accomplished using an interposition graft or a brachiocephalic vessel turndown, either in isolation or in combination with banding of the pulmonary artery. In most instances, repair of the VSD (with removal of the pulmonary artery band) would be undertaken some months later. Even after the introduction and successful achievement of one-stage repair of IAA and VSD closure, both via median sternotomy with cardiopulmonary bypass and hypothermic circulatory arrest, there persisted at many centers a bias favoring a staged approach to IAA with VSD or other more complex cardiac anatomy.35 In 1997, Mainwaring and Lamberti reported on their 10-year experience with a two-stage approach to repair IAA type B with VSD.36 Twenty-six of 27 patients survived stage 1 (arch repair with interposition graft). Twenty-two of 25 patients who underwent subsequent VSD closure were long-term survivors. Freedom from reoperation for arch graft enlargement was 86% at 3 years and 55% at 5 years.
In 1994, the CHSS published a report of their first multi-institutional outcome study of patients with interrupted aortic arch and ventricular septal defect.15 Patients were enrolled as neonates between 1987 and 1992, and 173 patients underwent reparative surgery. The initial procedure consisted of arch repair and VSD closure (one stage) in 116 (67%), arch repair and banding of the pulmonary trunk in 40, and only repair of the arch interruption in 17. Thus, one-stage repair was chosen in 67% of cases, with intent to undertake two-stage repair in 33%. In the past 2 decades, single-stage repair of IAA and intracardiac defects has gained wide acceptance. At most centers, it is the usual or routine technique used, with a two-stage approach being reserved for specific uncommon circumstances.37–41
Techniques of Arch Repair
An optimal method of arch repair would be one that could be performed safely and reproducibly in neonates, with minimal likelihood of stenosis in the short or long term. Thus, it would result in normal growth of all aortic segments including the anastomotic area. Ideally, it would not require use or division of any of the principal brachiocephalic vessels. In the traditional multistage approach, a synthetic tube graft was interposed between the proximal (ascending) and distal (descending) aortic elements. Although this can generally be accomplished without cardiopulmonary bypass support through either a lateral or an anterior approach, the certainty that the graft will not grow with the patient ensures an absolute requirement for arch re-intervention in all survivors.42–44 That re-intervention may consist of either graft replacement or augmentation in situ, or extra-anatomic placement of an additional graft. An alternative technique that has been used in both one-stage and two-stage strategies is the use of a turned-down brachiocephalic artery (carotid or subclavian artery) that is anastomosed to the descending aorta. Monro and colleagues published a small series demonstrating satisfactory growth of the arch elements and anastomotic region in the majority of patients at follow-up of 8 to 19 years.45 In the current era, the approach most often used is a one-stage approach entailing direct anastomosis between the ascending aorta and the descending aorta. This involves extensive mobilization of both proximal and distal elements. Several centers have reported excellent early outcomes with the technique of end-to-side anastomosis of the descending thoracic aorta either to the ascending aorta or to the ascending aorta and the underside of the proximal arch.41,46–48
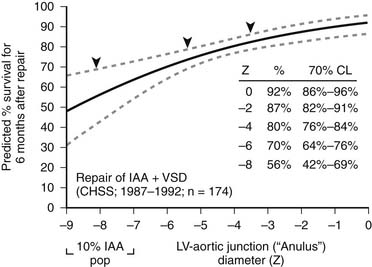
Patch augmentation of the anastomosis was described by Norwood in 1990, and intermediate-term results were reported by Jacobs and Norwood in 1995.49 This technique is intended to reduce tension on the anastomosis, to enlarge the connection between segments, and to address hypoplasia of the ascending aorta, which is particularly common in IAA types B and C.22,25 In addition, the use of patch augmentation generally obviates the need to divide either the left subclavian artery or an anomalous right subclavian artery to bring the elements of the arch together. It also lessens the likelihood of left bronchial compression, which can complicate some repairs of IAA.50 We have used cryopreserved pulmonary artery homograft tissue for the patch material. The 1994 CHSS report of outcomes in patients with IAA and VSD included an important observation concerning the potential benefit of patch augmentation of the amalgamation of the ascending and descending aorta in patients with additional levels of obstruction or hypoplasia in the left heart–aorta complex.15 Multivariable analysis identified subaortic or anular narrowing as incremental risk factors for death after repair (Fig. 122–2). The report by Jonas and associates concluded, “In the 20% of patients in whom obstruction existed elsewhere in the left heart-aorta complex, the percent survival was highest among those undergoing ascending aorta/arch augmentation.” More recently, in a CHSS analysis of intermediate- and long-term outcomes of an expanded cohort of 472 neonates with IAA, McCrindle and associates reported that re-intervention was more likely for those who had interrupted aortic arch repair by a method other than direct anastomosis with patch augmentation.17 In 2002, Roussin and associates described the incorporation of a pulmonary artery autograft patch into the aortic arch reconstruction.51
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


