Infective cardiomyopathies
Myocardial dysfunction is a well-recognized complication of viral, bacterial, fungal and parasitic infections. Worldwide, the most common infective myocarditis is Chagas’ disease caused by Trypanosoma cruzi, a protozoan organism endemic in rural areas of South and Central America. In the Western world, viral myocarditis caused by enterovirus and adenovirus infection is the most common cause of inflammatory heart muscle disease.
Viral myocarditis
Until quite recently, enterovirus (Coxsackie B) and adenovirus were the most commonly implicated viruses in acute myocarditis in adults and children.1–4 A causal association between enteroviruses and myocarditis was first suggested in studies showing a relationship between rising serum Coxsackie B antibody titers and acute symptomatic presentation.3 Subsequently, enteroviral genome has been detected in myocardial biopsy samples of patients with myocarditis and dilated cardiomyopathy using polymerase chain reaction techniques.4
More recently, studies suggest that the frequency of enteroviral and adenoviral infection in patients with clinically suspected myocarditis and dilated cardiomyopathy has reduced. Instead, parvovirus B19 infection, which usually causes erythema infectiosum (“slapped cheek” or “fifth” disease), seems more common in adults.5 Childhood parvovirus myocarditis has also been reported,6 including four cases of sudden cardiac death.7–10 Many other viruses have been implicated in myocarditis, including cytomegalovirus, hepatitis C virus, and herpes simplex virus11–14 (see Table 50.1).
Table 50.1 Studies showing viral genome persistence in patients with DCM or myocarditis
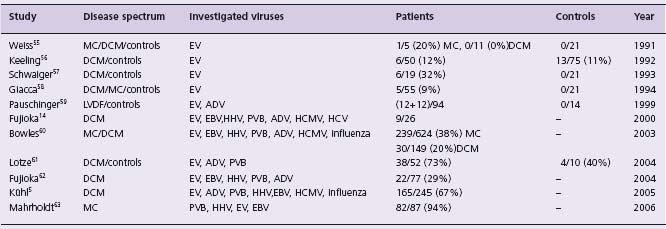
EV,enteroviruses (coxsackieviruses, polioviruses, echoviruses); ADV, adenoviruses; HHV, human herpes viruses; EBV, Epstein-Barr virus; HCMV, human cytomegalovirus; PVB, parvoviruses; DCM, dilated cardiomyopathy; LVDF, left ventricular dysfunction; MC, myocarditis.
HIV-related cardiomyopathy
The human immunodeficiency virus (HIV) infection affects an estimated 39.5 million living people worldwide, with 4.3 million new cases in 2006.15 Advances in treatment have substantially improved morbidity and mortality, but late cardiac complications are increasing as patients’ survival improves.16 These include myocardial dysfunction, pericardial effusion, endocarditis, malignancy and premature coronary disease. Observational data estimate the annual incidence of HIV-associated cardiomyopathy at 15.9 cases per 1000 HIV-positive patients.13 Among children with vertically transmitted HIV infection the 2-year incidence of congestive heart failure was 4.7%.17
Pathogenesis of the heart muscle dysfunction in HIV infection is still poorly understood. Opportunistic infections, malnutrition and antiviral treatment have been implicated.18 Some studies have also suggested a direct cardiotoxic effect of HIV and secondary effects caused by autoimmune responses against HIV and other cardiotropic viruses.12,19 This latter hypothesis is supported by the demonstration of a high frequency of circulating cardiac specific autoantibodies in HIV positive patients in comparison to normal controls.20
Clinical presentation and diagnosis
Presentation of infective myocarditis varies enormously. Malaise, low-grade fever, chest pain, transient electrocardiographic changes (T-wave changes, ventricular extrasys-toles), left ventricular dysfunction and biochemical signs of myocardial dysfunction are typical. In some cases, the clinical picture mimics acute myocardial infarction with ST segment elevation.21 Other cases present with fulminant heart failure,22 arrhythmias,23 conduction disturbance24 and sudden cardiac death.25
Although the echocardiogram is rarely entirely normal in myocarditis, the findings are non-specific. Evidence of impaired left ventricular systolic performance (with reduced fractional shortening and ejection fraction) is common. Regional wall motion abnormalities occur frequently. Left (or right) ventricular thrombus may be present. Evidence of left ventricular diastolic impairment may also be detected in myocarditis. Less often, right ventricular systolic and diastolic function may also be compromised. Echocardiography may show wall thickening caused by myocardial edema.26
Magnetic resonance imaging with gadolinium enahancement often demonstrates regions of signal enhancement.27,28 Mild to severe persistent perfusion defects using thallium-201 myocardial scintigraphy may be present in the acute phase, with partial recovery on follow-up.29
Routine blood tests such as full blood count and erythrocyte sedimentation rate are usually unhelpful in confirming or refuting the diagnosis of myocarditis. In a study of 80 patients with suspected myocarditis, 35% had elevated troponin T levels. Troponin T levels greater than 0.1 ng/mL had a specificity of 94% and a positive predictive value of 93%, although the sensitivity for detecting myocarditis was low at 53%.30 Similarly, troponin I has a high specificity (89%) for diagnosing myocarditis but a sensitivity of only 34%.31 Creatine kinase and its cardiac isoform CK-MB are less sensitive and specific than troponin, and are not clinically useful as a screening test in suspected myocarditis. Autoantibodies directed against myocardial proteins such as myosin and the adenine nucleotide translocator protein are reported in myocarditis, and correlate with progressive ventricular dysfunction.32,33
Endomyocardial biopsy (EMB) theorectically provides the means for establishment of a pathologic diagnosis.34 Only two clinical scenarios are class I indications: new-onset heart failure of less than 2 weeks’ duration associated with a normal sized or dilated left ventricle and hemodynamic compromise; and new-onset heart failure of 2 weeks’ to 3 months’ duration associated with a dilated left ventricle and new ventricular arrhythmias, second-or third-degree heart block, or failure to respond to usual care within 1 to 2 weeks. The rationale for these recommendations is that many patients presenting in this way have myocarditis and by determining the histologic type (lymphocytic, giant cell or necrotizing eosinophilic) prognostic information can be obtained and specific therapy initiated. The use of EMB to detect viral persistence is considered to be a research indication.
Management
General principles
General principlesofstabilization include afterload reduction, anticoagulation, diuresis and inotropic support. Patients with fulminant acute myocarditis may require intensive intravenous hemodynamic support. In severe cases, mechanical assist devices or extracorporeal membrane oxygenation may be necessary.35–38 Following initial stabilization, treatment should follow standard heart failure guidelines. Patients with left ventricular enlargement may require anticoagulation with warfarin. Patients with intractable and deteriorating heart failure may require cardiac transplantation.
Immunotherapy
(Table 50.2) Intravenous immunoglobulin has been extensively used in children with suspected myocarditis.39 Evidence for its use includes case reports,40,41 and case series42,43 of children and adults with acute myocarditis. However, only one double-blind randomized controlled trial of immunoglobulin has been conducted. In 62 patients with recent-onset heart failure and unexplained dilated cardiomyopathy, intravenous immunoglobulin failed to improve all-cause mortality or left ventricular ejection fraction.44 Furthermore, spontaneous improvement in left ventricular function occurred in both treatment and control groups.44 A recent Cochrane Database review concluded that current evidence does not support the use of intravenous immunoglobulin for the management of presumed viral myocarditis and that “intravenous immunoglobulin for presumed viral myocarditis should not be part of routine practice”45 (Class IIb, Level B).
Table 50.2 Studies or case reports of immunotherapy with IV gammaglobulin (IVGG) for myocarditis
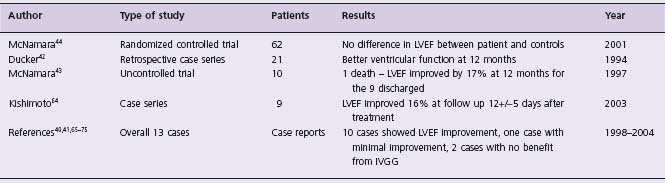
LVEF, left ventricular ejection fraction.
Immunosuppression
(Table 50.3) The hypothesis that immune responses to viral infection may cause the long-term sequelae of viral myocarditis has led to the use of immunosuppression to reduce the acute inflammatory response.1,46 A potential danger of this approach is that immunosuppression might impair the ability of the host immune system to eradicate virus.47 In mice, prednisolone increases mortality in animals with acute viral myocarditis.48 Observational unrandomized case series in children have reported a beneficial effect of immunosuppression in children with myocarditis.49–52 In the Myocarditis Treatment Trial, there was no difference in mortality or left ventricular function between the control group and the treatment groups (prednisolone plus either ciclosporin or azathioprine).53 In another study of patients with major histocompatibility complex expression on endomyocardial biopsy samples randomized to prednisolone and azathio-prine or placebo, there was improvement in left ventricular ejection fraction in the immunosuppressed group only, but no difference was observed in mortality, transplantation or rehospitalization rates over a 2-year follow-up period.54 On the basis of the available data immunosuppression therapy is considered as a Class IIa, Level C recommendation for children with myocarditis. For adults the data are Class IIb, Level B.
Table 50.3 Studies of immunosuppressive therapy for myocarditis
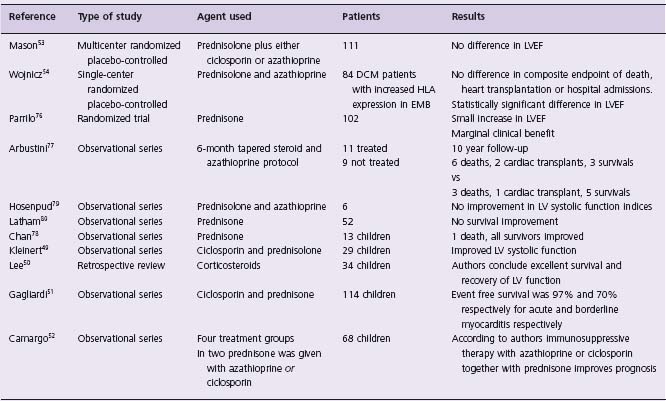
LVEF, left ventricular ejection fraction; DCM, dilated cardiomyopathy; HLA, human leukocyte antigen; EMB, endomyocardial biopsy.
Bacterial infections
Bacterial myocarditis is rare in the modern era but still occurs in high-risk groups such as immnunosuppressed patients. Pathogenetic mechanisms include direct bacterial invasion with myocardial tissue destruction, abscess formation and toxin-related cardiac dysfunction. Staphylococcus aureus is the most common cause of bacterial myocarditis producing cardiac dysfunction, rhythm disturbances, and myocardial rupture with secondary purulent pericarditis.81 Corynebacterium diphtheriae respiratory infections can proceed to myocardial involvement mediated by elaborated toxins and affecting predominantly the cardiac conducting system. Cardiac tuberculosis can produce miliary or nodular lesions82 and has been associated with severe heart failure,83 ventricular arrthymias84 and sudden cardiac death.85 Treponema whippleii causing intestinal lipodystrophy or Whipple disease may affect cardiac valves, pericardium and myocardium.86 Myocarditis can also be caused by Clostridium species.87 Brucella,88 Mycoplasma, Chlamydia,89 Salmonella90 and numerous other bacteria.
Therapy in the acute phase is supportive. Complete atrioventricular block should be treated with temporary or permanent transvenous pacing.91 Antitoxins are available and should be used when appropriate. Specialized antibiotic regimens directed against the pathogen usually resolve the infection.
Rheumatic carditis
Rheumatic carditis is a common manifestation of acute rheumatic fever following group A streptococcal (GAS) infection of the tonsillopharynx.92 The main pathogenetic process is a genetically predisposed autoimmune connective tissue damage, which affects primarily the cardiac valves.93
Diagnosis traditionally has been based on the constellation of symptoms and signs first introduced by Jones in 1944 and further updated in 1992.94 The presence of two major or one major and two minor manifestations together with evidence of antecedent streptococcal infection implies a high probability of acute rheumatic fever.
Rheumatic carditis is seen usually within 3 weeks of GAS infection. Prevalence was estimated around 50% of acute RF cases based on clinical findings; however, using modern echocardiographic tools this proportion is somewhat higher.95 Valvular dysfunction during the acute phase is typically seen as mitral and aortic valve regurgitation caused by ventricular dilation combined with leaflet restriction and thickening from active inflammation.95 The tricuspid valve can also be affected but the pulmonary valve is spared. Overt heart failure is usually the consequence of acute valvular regurgitation with no significant myocardial contractile dysfunction detected in several series.96,97 Parietal and visceral pericardial involvement with effusion is also rare. Moderate or severe carditis, recurrent rheumatic fever attacks or low educational level have been related to late valvular fibrosis and scarring causing debilitating hemodynamics and long-term morbidity.98
Cardiac troponin levels lie within the normal range, showing no myocardial necrosis despite the intense endo-cardial and connective tissue inflammation.99 Acute phase reactants, like C-reactive protein or erythrocyte sedimentation rate, are high, reflecting the rheumatic activity and helping to establish the diagnosis.
Treatment during the acute phase is supportive and antiinflammatory regimens with salicylates or steroids are dictated by the overall clinical picture. However, there is no convincing evidence supporting corticosteroid or immunoglobin administration during acute rheumatic carditis to prevent late valvular damage100 (Class III, Level B). Antibiotics (penicillin or erythromycin in penicillin allergy) are mandatory to eradicate the causative agent. Secondary prevention to avoid recurrences should be implemented in patients without carditis for 5 years or until the age of 18, in those with mild mitral regurgitation for 10 years or until the age of 25, and life-long prevention is suggested for severe valve disease and after valve surgery.101 However, the use of penicillin prophylaxis against recurrent rheumatic fever attacks has not always been efficient according to a recent Cochrane Database review (Class IIb, Level C). In the same review intramuscular penicillin injections every 2 – 3 weeks seem to be more effective than 4-week regimens or oral formulations.102 (Class IIa, Level C).
Rickettsial-spirochetal infections
Myocarditis caused by Coxiella burnetii infection may be encountered during the course of Q fever. This may present with precordial pain or heart failure symptoms. The prognosis is generally poor.103 Myocardial involvement with chest pain, elevated myocardial enzymes and depressed LV function has also been described with Rickettsia rickettsii infections, responsible for Rocky Mountain fever.104 Spiro-chetal infections including syphilis and Lyme disease (Borrelia burgdorferi) can also cause myocarditis. Apart from aortitis, myocardial involvement with left ventricular aneurysms has been reported in secondary syphilis.105 Bor-relia infection usually has neurologic, joint and cutaneous manifestations (erythema migrans) with cardiac involvement appearing within 3 weeks in approximately 8% of cases.106
Fungal infections
Fungi, as opportunistic pathogens, can cause myocarditis in immunodeficient patients receiving chemotherapy, steroids or immunosuppressive therapy as well as patients with malignancies. The micro-organism may spread from neighboring lung or mediastinal foci or by hematogenous dissemination. Among the various fungal species, cardiac aspergillosis has several case reports in patients with aquired immunodeficiency syndrome and is associated with poor prognosis.107
Parasitic infections
Parasitic infections can present with a wide spectrum of cardiac manifestations and remain a major public health issue in developing countries. Helminths or protozoa have the ability to survive and multiply in a variety of mammalian cells including cardiomyocytes, in spite of the host’s defensive immune mechanisms. This usually leads to chronic inflammatory responses that ultimately damage the heart muscle, conducting system and pericardial tissue. Chagas’ disease, amebiasis, toxoplasmosis, cysticercosis, echinococcosis and trichinellosis are all parasitic infections that commonly affect the heart.108 For a review of Chagas’ disease see Chapter 51.
A number of genetic and acquired disorders result in myo-cardial infiltration or excessive storage of various substances. Most are associated with extracardiac manifestations and often present exclusively in childhood. A relatively small number present with predominantly cardiac involvement in adults, most notably amyloidosis, hemochromatosis, and Anderson–Fabry disease.
Amyloidosis
Amyloidosis is caused by extracellular deposition in various tissues of proteinaceous material with a characteristic cross-beta sheet quaternary structure, in which strands from different monomers are aligned perpendicular to the axis of the fibril.109 Amyloid deposits are characterized by a change in the fluorescence intensity (apple green birefringence) under polarized light microscopy of planar aromatic dyes such as Congo red and stain a characteristic color with sulfated alcian blue. The classification of amyloidosis is based on the different protein precursors that make up the amyloid fibrils. Amyloid protein deposition occurs in atria, ventricles, coronary vessels, conduction system and valves. The degree of cardiac involvement varies between each subtype. Hematologic disorders associated with excessive light chain (AL) immunoglobulin production are the most common cause of cardiac amyloid. Familial forms caused by the accumulation of mutant proteins (transthyretin or A-apolipoprotein) have variable cardiac involvement. Secondary amyloidosis, due to deposition of serum amyloid A protein associated with chronic inflammatory diseases, rarely affects the heart to a clinically significant degree.
AL amyloid
Deposition of clonal immunoglobin light chains (with or without multiple myeloma) is the most common cause of amyloidosis. Cardiac involvement occurs in up to 50% of patients with AL amyloid, but clinically isolated cardiac disease is seen in less than 5% of patients.110 Myocardial wall thickening caused by extracellular deposits without myocyte hypertrophy results in stiff non-dilated ventricles and biatrial dilation.111 Progressive dyspnea and profound fluid retention dominates the clinical picture. Low cardiac output causes weakness, fatigue and exercise intolerance. Typically, heart failure caused by cardiac infiltration is rapidly progressive with a median survival of less than 1 year.112
Chest discomfort or angina pectoris is common, but coronary angiography usually reveals normal epicardial arteries. Perivascular infiltrates and evidence of impaired myocardial flow reserve suggest small vessel disease as the underlying cause.113 Obstructive intramural coronary amy-loidosis has also been reported in autopsy series.114 Small but persistent cardiac troponin elevations reflecting subtle ongoing myocardial necrosis are also reported.115
Syncope is caused by impaired cardiac output, arrhythmia and amyloid autonomic neuropathy.116 Symptomatic supraventricular arrhythmias associated with atrial enlargement are frequent.117 Infiltration of the atria may add to the risk for atrial fibrillation.118 Infiltration of the sinus node and atrioventricular conduction system causes bradyarrhythmia and complete heart block.119 Documented complex ventricular arrhythmias are rare, but may underlie some cases of sudden death; sudden death in advanced heart failure cases may also be caused by electromechanical dissociation.120
Cardiomegaly and pulmonary congestion with bilateral pleural effusions are seen at late stages on chest radiography. Cardiac troponins and B-type natriuretic peptide reflect the severity of cardiac involvement121 and are associated with poor prognosis.115,122 The resting 12-lead electrocardiogram (ECG) usually demonstrates low voltages (< 5 mV in limb leads) or pseudo-infarction patterns with poor right precordial R-wave progression or inferior Q-waves.123 Criteria for left ventricular hypertrophy are uncommon, probably because myocardial wall thickening is due to extracellular infiltration rather than true hypertrophy.124 Atrial fibrillation occurs in 7 – 25% of patients and poses a significant risk of thromboembolism.109 Ambulatory ECG monitoring reveals ventricular ectopics, couplets, and non-sustained ventricular tachycardia. Complex ventricular arrhythmias are considered harbingers of lethal arrhythmic events.125 Sinus node dysfunction, supraven-tricular arrhythmias and atrioventricular conduction abnormalities may be evident.119 Late potentials on signalaveraged ECG are common in patients with cardiac amyloidosis and predict sudden death independently of echocardiographic disease markers.126 Electrophysiology studies may reveal subtle abnormalities of the His – Purkinje system not evident on surface ECG. HV prolongation is common, and is of prognostic significance in some series.127
Echocardiography typically shows that global indices of systolic function are preserved until the final stages of the disease. However, decreased longitudinal deformation is common and precedes the onset of heart failure symptoms128 Two-dimensional echo findings in advanced cardiac amyloidosis are characteristic.129 Increased biventricular wall thickness with concentric distribution is typical, although asymmetric septal hypertrophy with normal ejection fraction can be encountered. Dynamic left ventricular outflow tract obstruction is extremely unusual. Cardiac valves appear infiltrated and thick, the atria are dilated and the interatrial septum is often thickened. A small pericardial effusion is also common. The peculiar, granular texture of the affected myocardium has traditionally been described as an amyloid feature, but is difficult to quantify without digital image analysis. Transmitral Doppler recordings in advanced cases demonstrate a restrictive left ventricular filling pattern. Diastolic dysfunction indices are better prognostic markers than left ventricular wall thickness.130
Other imaging modalities such as cardiac magnetic resonance imaging are used to further assist morphologic description and tissue characterization. Subendocardial late gadolinium enhancement is considered to reflect the transmural distribution of amyloid deposits and cardiac amyloid load.131 Abnormal gadolinium kinetics are also described, although the sensitivity of this observation is uncertain. Nuclear scans with123I-labeled serum amyloid P component are highly specific. The isotope localizes selectively in amyloid infiltrates and aids the diagnosis.132 Other scintigraphic techniques have been used to demonstrate cardiac adrenergic denervation even before the development of clinically apparent heart disease.133
The diagnosis of amyloidosis requires a tissue biopsy. This need not be from the heart if the echocardiographic appearances are typical for cardiac amyloidosis. Fine-needle aspiration of the abdominal fat is positive for amyloid deposits in over 70% of patients with AL amyloidosis. If negative, endomyocardial biopsy has a very high sensitivity. Once confirmed, all patients with AL amyloid should be assessed for a plasma cell dyscrasia.
Management of cardiac amyloidosis is directed against symptoms of heart failure. Specific therapies designed to impede precursor production and fibril formation should be implemented whenever possible.134 Diuretics, often in high dose, are the cornerstone of the palliative heart failure regimen. ACE or angiotensin II inhibitors should be introduced with extreme caution as they are often poorly tolerated and there are no data on their efficacy in cardiac amyloid. Aldosterone inhibitors might be helpful in advanced cases. Sensitivity to digoxin, attributed to enhanced drug binding on amyloid fibrils, demands cautious administration.135 Although there are no data, the presence of atrial fibrillation in AL amyloidosis should always prompt anticoagulation with warfarin because of a very high rate of thromboembolism.
Management of the underlying amyloid disease process is based mainly on single-center experience with few randomized studies. In AL amyloidosis, progression of systemic disease contributes substantially to poor outcomes even when heart transplant is provided.136 Heart transplantation followed by high-dose chemotherapy in combination with stem cell transplantation, is promising with short-to intermediate-term survival being now similar to patients receiving cardiac transplant for other indications.137
Familial amyloid
Hereditary amyloidosis is most commonly transmitted as an autosomal dominant trait with high penetrance, caused by mutations in the gene encoding transthyretin (TTR). More than 70 mutations in the transthyretin gene have been reported as amyloidogenic but one of the most common is a substitution of isoleucine for valine at position 122 which occurs in 4% of the black US population.138 TTR amyloidoses usually present after the third decade of life with polyneuropathy and cardiomyopathy. Sometimes cardiac involvement is clinically confined to atrial arrhythmia and conduction disease. Despite significant myocardial infiltration, heart failure symptoms are less profound and survival is much better than AL amyloidosis.139 Toxic light chain effects, absent in TTR amyloidosis, may explain the different clinical outcome.140
As transthyretin is produced by the liver, the definitive treatment of TTR amyloidosis is liver transplantation. Unfortunately, myocardial wall thickening progresses in some patients with TTR amyloid cardiomyopathy following liver transplantation, probably due to the continued deposition of wild-type transthyretin in the myocardium. Combined liver and heart transplantation or heart transplantation alone has been performed for TTR amyloidosis in small numbers. In vitro studies suggests that non-steroidal agents such as diflunisal can stabilize transthyretin.141
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


