Hypoplastic Left Heart Syndrome
Peter J. Gruber
Thomas L. Spray
INTRODUCTION
Hypoplastic left heart syndrome (HLHS) comprises a wide spectrum of anatomic abnormalities with the common features of left ventricular hypoplasia and hypoplasia of the ascending aorta. At one end of the spectrum, there may be some mild left ventricle hypoplasia, mild aortic stenosis, and aortic coarctation. At the other end of the spectrum, however, there is complete absence of the left ventricle, aortic atresia, and aortic arch hypoplasia or even interrupted aortic arch.
HLHS is a uniformly fatal disease if untreated. It represents 5% of all congenital heart disease and is responsible for nearly 25% of cardiac deaths in the first week of life. Of 10,000 live births, approximately 1.8 will be born with HLHS, with a slight male predominance. Of these, 25% will also have a noncardiac anomaly and 5% a chromosomal abnormality (trisomies 13, 18, and 21). Syndromic lesions are rare, with Turner syndrome (monosomy X) the most common. The recurrence risk is 2.2% for one affected sibling and 6% for two affected siblings, suggesting some genetic predisposition but arguing against a simple effect.
Surgery for HLHS is one of the great successes in the management of congenital heart disease. Before the 1980s, HLHS was a uniformly lethal condition. However, over the last 25 years the repair of HLHS has become a standard operation in nearly all institutions. In 1952, Lev first described maldevelopment of the left-sided cardiac structures in combination with a small ascending aorta and transverse arch. By 1958, Noonan and Nadas had further defined the syndrome to describe a variety of cardiac malformations of left heart structures. The first report of any attempt to palliate a patient with mitral atresia was by Redo in 1961, who performed an atrial septectomy using inflow occlusion through a right thoracotomy; the patient died soon after the operation. In 1968, Sinha outlined the management principles still in use today that include creation of an unobstructed atrial communication, unrestricted ductal flow, and control of pulmonary blood flow. Cayler described an anastomosis between the right pulmonary artery (RPA) and ascending aorta with banding of both right and left pulmonary arteries (LPAs). It is of interest that 35 years later, pulmonary artery (PA) banding is being used in certain centers for selected children who present with a medical or anatomic situation unsuitable for stage 1 Norwood reconstruction; this first-stage hybrid procedure involves stenting the ductus arteriosus and atrial septal defect and using bilateral PA bands. Litwin, Mohri, and others performed operations that were variations of the principles of palliation that were unsuccessful but contributed to the development of the knowledge of the disease and its repair. In 1977, Doty described primary reconstruction that included atrial septation and a right atrium (RA)-to-PA Fontan circuit. Again, although no patients survived, this experience established the principle that one-stage reconstruction with a Fontan repair would not be successful due to high neonatal pulmonary vascular resistance. Levitsky, Behrendt, and others described multiple variations of surgical procedures that, although they demonstrated no long-term success, established the principle of staged reconstruction with initial palliation followed by later separation of the systemic and pulmonary circulations. However, it was Norwood who in 1980 first achieved successful palliation in infants. In 1983, he described the first successful staged approach culminating in a Fontan repair. Today, the Norwood procedure remains the primary reconstructive approach.
 ANATOMY
ANATOMYPatients with HLHS are currently categorized on the basis of atrioventricular (AV) and semilunar valvular morphology into three primary subsets: (1) aortic atresia with mitral atresia (40%), (2) aortic stenosis with mitral stenosis (30%), and (3) aortic atresia with mitral stenosis (30%) (Fig. 96.1).Aortic stenosis with mitral atresia is rare. HLHS variants include malaligned AV canal, double-outlet right ventricle with mitral atresia, tricuspid atresia with transposed great arteries, and univentricular heart with aortic stenosis. There is frequent leftward and posterior deviation of the septal attachment of the septum primum, but this feature is unlikely to be a causal developmental mechanism because it is commonly seen in other congenital heart disease phenotypes. Usually, the superior vena cava (SVC) and inferior vena cava (IVC) are normally connected to the RA, although in about 15% of patients a left SVC-to-coronary sinus is present. Other structural abnormalities of the heart are rare, with <5% of patients demonstrating AV valvular dysplasia. Also, rare in nonsyndromic forms (<5%) are abnormalities of pulmonary venous return or an interrupted aortic arch. Abnormalities in brain development are increasingly associated with children with severe congenital heart disease, and these may be a high-risk group for operative repair. The pulmonary vascular tree is also abnormal, with an increase in number of vessels as well as muscularity.
The developmental mechanisms that underlie HLHS are obscure from a molecular standpoint because there are no mutations that have been robustly associated with this condition. Despite the existence of rare family clustering of HLHS, linkage analysis has been largely unproductive. Indeed, it is likely that there exists considerable genetic heterogeneity in this as well as other CHD phenotypes. However, embryologically, there are clues. The severe hypoplasia of left heart structures may be a consequence of limited flow during development secondary to a primary abnormality of either left ventricular inflow or left ventricular outflow. Primary defects of myocardial growth are unlikely to be a mechanism for this disease because the myocardium appears normal. In addition, approximately 5% of patients with aortic atresia demonstrate an unrestrictive ventricular
septal defect, and in such cases, there is nearly always normal development of the left ventricle and mitral valve.
septal defect, and in such cases, there is nearly always normal development of the left ventricle and mitral valve.
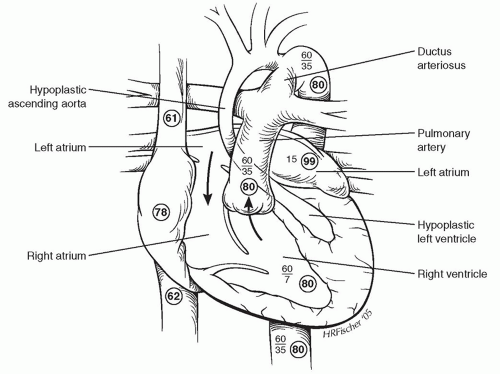 Fig. 96.1. Anatomic features and representative hemodynamic parameters for unrepaired hypoplastic left heart syndrome. Oxygen saturations are enclosed in circles, and blood pressures are indicated by standard nomenclature. |
PRESENTATION AND INITIAL MANAGEMENT
The normal fetus has a parallel circulation that adequately supports single-ventricle physiology before birth. Three communications (the ductus venosus, foramen ovale, and ductus arteriosus) shunt oxygenated placental blood largely past the hepatic and pulmonary beds to supply the splanchnic circulation. HLHS is well supported in this physiology and is unlikely a cause of fetal demise. More likely, it is a secondary result of early obstructive lesions of either mitral or aortic valvular development. This has been supported in animal models of either mitral or aortic stenosis with resulting left ventricular hypoplasia. However, the primary cause of the obstructive flow lesion leading secondarily to HLHS is unknown. There are no known genetic animal models that fully recapitulate HLHS despite the existence of a large number of mutations that affect valvular development. This argues either for a complex early event that is the result of multiple factors or an early insult influence by subsequent modifiers.
The presentation of infants with congenital heart disease has changed dramatically over the last 15 years. In most large centers, the majority of patients are identified through prenatal echocardiography, although this early identification has not consistently correlated with better outcome. Although some tachypnea and mild cyanosis may be present, it is not until the ductus arteriosus begins to close that the children exhibit impaired systemic perfusion with pallor, lethargy, and diminished femoral pulses. Cardiac examination reveals a dominant right ventricular impulse, a single-second heart sound, and often a nonspecific soft systolic murmur. Electrocardiogram examination reveals right atrial enlargement and right ventricular hypertrophy. Chest X-ray occasionally shows mild cardiomegaly and increased pulmonary vascular markings.
Physical examination of children with HLHS usually appears normal. The examination is determined by the underlying anatomy as well as the duration of the disease. Poor perfusion, weak distal pulses (that may or may not be present, depending on the size of the ductus), acidosis, and a sepsis-like picture may all confound the diagnosis. In the absence of risk factors or laboratory findings consistent with sepsis, one should search for left-sided obstructive lesions. There are no specific laboratory indicators of HLHS, and most patients usually exhibit normal values. With ductal closure and malperfusion, end-organ compromise may be reflected by altered hepatic and renal function tests.
Many mothers of fetuses with HLHS will have had a fetal echocardiogram at 20 weeks with reasonable visualization of cardiac structures. It is neither feasible nor cost effective to screen all pregnancies; therefore a selective approach is taken in which only those mothers at high risk are screened. Frequently, a ventricular size discrepancy is the first hint of impending problems. Certainly, the presence of an intact or restrictive atrial septum with HLHS should prompt term high-risk delivery in an institution in which an urgent postdelivery palliation can be performed safely and rapidly. Emergent operative atrial septectomy is poorly tolerated. The use of prenatal screening improves the prenatal condition of the child but may not improve outcome (at least in cases of transposition of the great arteries or HLHS). After delivery, the infant should undergo two-dimensional and Doppler echocardiography, which defines the anatomy sufficiently for medical and surgical decision-making. It is important to distinguish HLHS from other diseases that may mimic certain features. Chest radiography often demonstrates mild cardiomegaly and excessive pulmonary blood flow. Head ultrasound should be obtained in all patients to rule out intracranial hemorrhage and minimize the risks of heparinization and potential circulatory arrest. Patients with medical necrotizing enterocolitis should ideally have a 7-day course of intravenous antibiotics before repair if they are hemodynamically stable. Preoperative stabilization is critical to the ultimate outcome of patients with HLHS regardless of anatomic subtype, though operation should not be substantially delayed. Nearly all patients with suspected HLHS are transported on prostaglandin E1 at a dose of 0.01 to 0.025 µg/kg/minute. Two clinically important dose-dependent side effects of prostaglandin E1 infusion are hypotension and apnea, although these are infrequent. Umbilical arterial and umbilical venous lines are used for central access in most patients. Most patients can ventilate with a natural airway and demonstrate more favorable hemodynamics while extubated. Supplemental oxygen should generally be avoided as it acts as a pulmonary vasodilator, decreasing pulmonary vascular
resistance, increasing the ratio of pulmonary-to-systemic blood flow, and thus decreasing systemic perfusion. Inotropic support is rarely necessary, although it may be required for support in patients who have suffered a perinatal insult. The goal of these maneuvers is to get the patient to the operating room in as stable condition as is possible.
resistance, increasing the ratio of pulmonary-to-systemic blood flow, and thus decreasing systemic perfusion. Inotropic support is rarely necessary, although it may be required for support in patients who have suffered a perinatal insult. The goal of these maneuvers is to get the patient to the operating room in as stable condition as is possible.
 SURGICAL THERAPY
SURGICAL THERAPYThe primary therapy for HLHS is staged reconstructive surgery leading to a modified Fontan-Kreutzer procedure. Over the last 25 years, the Norwood procedure has evolved and is now a standard operation in nearly all institutions for HLHS. There are three primary goals of stage I palliation: (1) establishment of unrestricted interatrial communication to provide complete mixing and avoid pulmonary venous hypertension, (2) establishment of a reliable source of pulmonary blood flow, allowing pulmonary vasculature development and minimizing the volume load on the single ventricle, and (3) providing unobstructed outflow from the ventricle to the systemic circulation.
We offer surgical palliation to nearly all patients with HLHS, including very low birth weight infants and those with nonlethal genetic syndromes. In certain complicating situations, primary transplantation (or other forms of palliation described below) may be considered, such as in severe aortic or AV regurgitation, or dilated cardiomyopathy.
Stage I Reconstructive Surgery
The child is brought to the operating room and ventilated on room air, with care taken to avoid hyperventilation. A full midline sternotomy is performed and a sternal retractor placed. The thymus is removed in its entirety, with care being taken to avoid the phrenic nerves. The pericardium is opened, and an obligatory mediastinal inspection is performed to confirm the echocardiography, especially to identify the abnormalities of the aortic arch and coronary arteries as coronary orientation may alter the surgical approach. The ascending and descending aorta, brachiocephalic vessels, ductus arteriosus, and PAs are extensively mobilized, with care taken to avoid damage to the recurrent laryngeal nerve. No attempt is made to dissect the systemic veins. Purse-string sutures are placed in the proximal main pulmonary artery (MPA) and generously around the right atrial appendage, through which heparin is administered. A previously thawed homograft pulmonary hemipatch is then trimmed in an extended arrowhead shape and set aside (Fig. 96.2A). Two perfusion techniques are commonly used for operative repair: deep hypothermic circulatory arrest (DHCA) and selective antegrade continuous cerebral perfusion. Despite intensive investigation, there is no consensus regarding the superior approach. After the activated clotting time reaches 300 seconds, the patient is cannulated with the arterial cannula at the base of the MPA and a single venous cannula in the RA. Cardiopulmonary bypass is initiated and tapes brought down around the branch PAs. The patient is cooled to 18°C over 15 minutes, during which time any remaining dissection is performed. During this period of cooling, a side-biting clamp is placed on the innominate artery, and a polytetrafluoroethylene (PTFE) graft (usually 4.0 mm for patients >3.2 kg and 3.5 mm in smaller infants) is anastomosed in an end-to-side manner. The clamp is removed and flow assessed. If blood does not briskly flow from the open shunt, the anastomosis should be revised. A large hemoclip is placed to temporarily occlude the shunt. On initiation of circulatory arrest, tapes are brought down around the brachiocephalic vessels and a vascular clamp is placed on the descending aorta distal to the ductal insertion site. Cardioplegia is administered antegrade through a side port on the arterial cannula. After draining the patient of blood, all cannulas and PA tapes are removed. The ductus arteriosus is ligated on the PA side and divided on the aortic side. The atrial septum is completely excised working through the atrial purse string (Fig. 96.2B). Visualization can be improved through a right atriotomy, although this is seldom necessary. Next, the MPA is divided close to the branch PAs, and the defect in the distal MPA segment is closed either with an oval homograft patch or primarily in a vertical manner.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


