Hypertrophic Cardiomyopathy
William J. McKenna
Perry M. Elliott
Introduction
Hypertrophic cardiomyopathy (HCM) is a phenotypically heterogeneous condition that affects individuals of all ages. The disease is typically inherited in an autosomal dominant fashion with variable clinical penetrance. In adults, the majority of cases are caused by mutations in one of 12 cardiac sarcomeric protein genes; in infants and children, left ventricular hypertrophy is often associated with congenital malformations and syndromes, inherited disorders of metabolism, and neuromuscular diseases.
The disease is characterized by myocardial hypertrophy, most commonly affecting the interventricular septum, disorganization (“disarray”) of cardiac myocytes and myofibrils, myocardial fibrosis, and small-vessel disease. Twenty-five percent of patients have resting left ventricular outflow tract obstruction.
Patients present throughout life with chest pain, dyspnea, palpitations, and syncope; the most important complication of the disease is sudden death, which occurs with an annual incidence of approximately 1%. Other complications include atrial arrhythmia, thromboembolism, infective endocarditis, and congestive cardiac failure. Symptoms can be improved with pharmacologic therapy or relief of left ventricular outflow tract obstruction using alcohol ablation or surgery; cardiac transplantation is required in a small minority of cases. Sudden death prevention is based on the identification of clinical risk markers; patients with multiple risk factors are treated with implantable cardioverter defibrillators.
Historical Perspective
Asymmetric hypertrophy of the interventricular septum was first described in 1869 by Liouville (1) and Hallopeau (2), but it was only in the 1950s that hypertrophic cardiomyopathy was established as a discrete clinical entity with the description of “functional obstruction of the left ventricle” by Sir Russell Brock (3) and asymmetric septal hypertrophy by Donald Teare (4). There followed a period of intense clinical investigation in which the characteristic morphologic and hemodynamic features of the disease were defined. In the 1960s (5,6,7,8,9), reliance on stethoscope and cardiac catheterization meant that left ventricular outflow tract obstruction was considered to be the diagnostic hallmark of HCM, a fact reflected in the many pseudonyms for the disease used during this period (e.g., idiopathic hypertrophic subaortic stenosis [IHSS]); in the 1970s, M-mode echocardiography (10,11,12) reemphasized asymmetric septal hypertrophy (ASH) as the diagnostic hallmark of the disease, but the advent of two-dimensional echocardiographic imaging demonstrated that virtually any pattern of unexplained myocardial hypertrophy is consistent with the diagnosis (13,14).
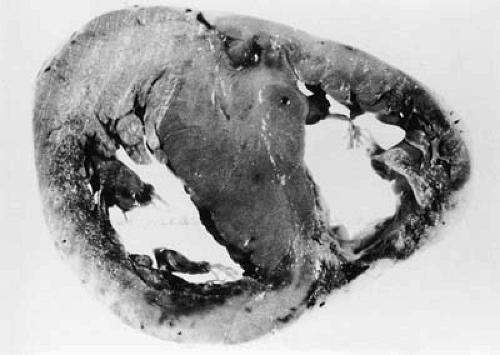 FIGURE 29.1. Myocardial section demonstrating the classic appearance of severe asymmetric septal hypertrophy in a patient with hypertrophic cardiomyopathy. (From Teare D. Asymmetrical hypertrophy of the heart in young adults. Br Heart J 1958;20:1–8. ) |
In the 1980s, work on the clinical pathophysiology of the disease continued, but the growing recognition that the majority of cases were familial led to the identification of the first genetic mutation in the gene encoding the β-myosin heavy chain (15). Since then, numerous mutations in this and other sarcomeric proteins have been reported, leading to the modern concept of the disease as a disorder of the myocyte contractile apparatus. This remains the current view, but genetic characterization has illustrated the diversity of clinical expression and is beginning to show that similar mutations can cause dilated and restrictive cardiomyopathy in the absence of hypertrophy (16,17).
Pathology
Macroscopic
Hypertrophic cardiomyopathy (HCM) is defined by the presence of unexplained myocardial hypertrophy (18,19), typically involving the interventricular septum more than the posterior or free wall of the left ventricle (4,5,6,7,8,9,10,11,12,13,14,19,20,21) (Fig. 29.1). Hypertrophy is less commonly concentric and can be confined to the left ventricular apex (19,20,21,22,23); right ventricular hypertrophy is also common but has not been reported in isolation (24). The cut surface of involved myocardium has a characteristic appearance with abnormally short and broad muscle fibers running in different directions. A patch of subendocardial thickening on the septum caused by repeated contact with the anterior leaflet of the mitral valve may be seen in patients who have had outflow tract obstruction (20,21).
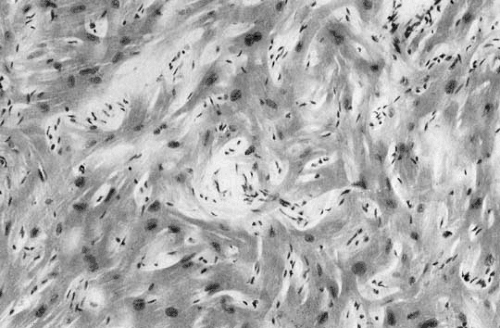 FIGURE 29.2. Myocyte disarray. Myocytes cross at right angles and display abnormal intercellular connections. Myocyte nuclei are large and of different sizes. There are foci of fibrosis containing spindle-shaped connective tissue cells. There is also evidence of marked disorganization of the intracellular myobrillar structure. Hematoxylin and eosin stain. (Courtesy of Prof. M. Davies, St. George’s Hospital Medical School, London.) |
Microscopic
Microscopically HCM is characterized by myocyte disarray (18,19,20,21,25,26,27) (Fig. 29.2), in which there is loss of the normal parallel arrangement of cardiomyocytes, cells instead forming circles or whorls around foci of connective tissue. The myofibrillar architecture within cells is also disrupted (myofibrillar disarray). Individual myocytes vary in shape and have pleomorphic nuclei and abnormal intercellular connections (28). Myocyte disarray is not specific to HCM, and it occurs in other syndromic causes of left ventricular hypertrophy such as Noonan syndrome and Friedreich ataxia as well as congenital heart disease, hypertension, and aortic stenosis (20,21,25,26,27,29,30). In HCM, it is typically more extensive, occupying 20% or more of at least one ventricular tissue block and more than 5% of total myocardium at postmortem. Disarray is often associated with myocardial fibrosis in the form of extensive scarring or a more diffuse interstitial pattern (20,21,25,26,27,29,30,31,32,33). Abnormal small intramural arteries are common, particularly in patients with ventricular dilation and reduced systolic function (20,21,27,34,35,36) (Fig. 29.3).
Molecular Genetics
Many cases of left ventricular hypertrophy in infants and young children are associated with congenital malformations and syndromes, inherited metabolic disorders, and neuromuscular diseases (Table 29.1) (18,19,26,29,30,37,38,39,40,41,42,43). The majority of adolescents and adults with HCM have familial disease with an autosomal dominant pattern of inheritance; approximately 50% to 60% of patients have mutations in one of eight genes that encode different components of the cardiac sarcomere (Fig. 29.4): β-myosin heavy chain (chromosome 14); cardiac troponin T (chromosome 1); cardiac troponin I (chromosome 19);
α-tropomyosin (chromosome 15); cardiac myosin-binding protein C (chromosome 11); the essential and regulatory myosin light chains (chromosomes 3 and 12, respectively); and cardiac actin (chromosome 15). Mutations in three other sarcomeric protein genes have been reported: titin (62), troponin C (63), and α-cardiac myosin heavy chain (19,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62). Some adult patients with apparently unexplained ventricular hypertrophy have diseases such as Anderson-Fabry disease (63), mitochondrial disease (64), and a rare phenotype comprising HCM, Wolff-Parkinson-White syndrome, and premature conduction disease caused by mutations in the gene encoding the γ-subunit of AMP kinase, an important regulator of cellular energy homeostasis (65,66,67). Other examples of nonsarcomeric gene mutations that can result in HCM phenocopies include LAMP-2 (Danon disease) (67,68), human muscle LIM protein (69), and phospholamban promoter (70).
α-tropomyosin (chromosome 15); cardiac myosin-binding protein C (chromosome 11); the essential and regulatory myosin light chains (chromosomes 3 and 12, respectively); and cardiac actin (chromosome 15). Mutations in three other sarcomeric protein genes have been reported: titin (62), troponin C (63), and α-cardiac myosin heavy chain (19,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62). Some adult patients with apparently unexplained ventricular hypertrophy have diseases such as Anderson-Fabry disease (63), mitochondrial disease (64), and a rare phenotype comprising HCM, Wolff-Parkinson-White syndrome, and premature conduction disease caused by mutations in the gene encoding the γ-subunit of AMP kinase, an important regulator of cellular energy homeostasis (65,66,67). Other examples of nonsarcomeric gene mutations that can result in HCM phenocopies include LAMP-2 (Danon disease) (67,68), human muscle LIM protein (69), and phospholamban promoter (70).
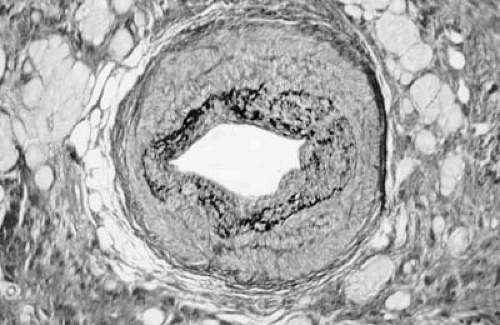 FIGURE 29.3. Section through a small artery demonstrating smooth muscle disorganization in the intima and media, resulting in an abnormally high ratio of medial width to lumen. In the adjacent myocardium there is extensive replacement of myocytes (yellow) by fibrous tissue (red). Elastica van Gieson stain. (Courtesy of Prof. M. Davies, St. George’s Hospital Medical School, London.) |
Pathophysiology of Sarcomeric Protein Gene Mutations
The proteins of the sarcomere, the functional unit of myocyte contraction, are organized into thick filaments (myosin heavy chain, myosin-binding protein C, and myosin essential and regulatory light chains) and thin filaments (actin, troponins T, I, and C, and tropomyosin) (44,45,46,71,72,73) (Fig. 29.4). Myocyte contraction occurs as the myofilaments slide over each other in a repetitive cycle of myosin and actin attachment, energy-dependent conformational changes in myosin, and actin–myosin release. The globular head of myosin heavy chain, connected through a flexible hinge region to the rod, contains enzymatic activity for ATP hydrolysis and actin-binding sites. Contraction is initiated following a rise in cytosolic calcium, which then binds the troponin complex and α-tropomyosin; this releases the inhibitory effect of troponin I on actin–myosin interaction, allowing actin to bind tightly to the myosin head; ATP then binds myosin, altering the conformation of the actin-binding sites and displacing the myosin head along the thin filament. ATP hydrolysis and release of ADP and Pi results in force generation (the power stroke) and restoration of the unbound conformation of myosin (44,45,46,72). Force transmission to the myocyte cytoskeleton is mediated via a complex of molecules including cardiac myosin-binding protein C, titin, dystrophin, and the sarcoglycan complex (44,45,46,73).
TABLE 29.1 Causes of Left Ventricular Hypertrophy in Children and Adults | |
|---|---|
|
Mutations in genes encoding sarcomere proteins might interfere with sarcomeric function in a number of ways (44,45,46;71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95). Inactivation of an allele can result in a reduced amount of functional protein (haploinsufficiency), whereas others produce a mutant protein that interferes with normal protein function (dominant negative) or has a novel function. Animal models of mutated myosin heavy chain (75,83,87,94), myosin-binding protein C (76,84), and troponin T (78,82) mimic some aspects of human disease, including cardiac hypertrophy, myocyte disarray, and interstitial fibrosis (74). Most transgenic models suggest that HCM results from the dominant negative effects of mutant proteins on sarcomere function and not haploinsufficiency or altered stoichiometry of sarcomere components. In addition, there seems to be a dose response, the levels of mutant sarcomere protein expressed within the heart correlating with myocyte dysfunction (76). Some effects
of human hypertrophic mutations relate to their proximity to critical binding sites; for example, in the myosin head domain, where affected residues interfere with the actin-binding site or the nucleotide (ATP)-binding pocket (71,72,73,74,75



of human hypertrophic mutations relate to their proximity to critical binding sites; for example, in the myosin head domain, where affected residues interfere with the actin-binding site or the nucleotide (ATP)-binding pocket (71,72,73,74,75
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
