High-Altitude Physiology and Clinical Disorders
INTRODUCTION
Altitude physiology typically focuses on people above 2500 m; ~8000 ft. Altitudes above that are sometimes subdivided into very high (3500–5500 m; ~11,500–18,000 ft) and extreme (>5500 m; >18,000 ft). An estimated 40 million people travel each year to altitudes >2500 m (~8000 ft),1 and as many or more travel to altitude for leisure and sports, and work in mines, military or border operations, and the like. Altitude medicine considers the clinical disorders associated with acclimatization by the travelers, workers and migrants, and with adaptation by people with lifetimes or populations with millennia of residence (an estimated 83 million people).2
With a hurried ascent, many (~80%) will report a transient headache (high-altitude headache or [HAH]), and some will develop one of three forms of acute high-altitude illness: acute mountain sickness (AMS) and HAH, high-altitude cerebral edema (HACE), and high-altitude pulmonary edema (HAPE).3,4 AMS (see Table 92-1) and HAH are annoying and interfere with activity and work, however, HACE and HAPE can be fatal with mortality rates approaching 30%.5–7 Among some residents, chronic mountain sickness (CMS) and right ventricular hypertrophy develop over months to years of residence at altitude. Birth weights are generally lower and the rate of small-for-gestational-age babies and congenital heart defects are higher than that in lowland populations.4
TABLE 92-1 Lake Louise Symptom Score Self-Report Questionnaire. Fill Out Before and Each Morning Upon Exposure to Altitude or on a Trek
Hypoxemia (FIO2 equivalent to ~17% O2 at 2500 m, down to ~8% O2 at the summit of Everest) causes the physiologic responses and illnesses. Altitude-related exposure to cold and extreme exercise may also contribute to illness. Other environmental features may include UV radiation, trauma, and infection that are not covered in this chapter. Finally, dealing with illness in a remote area can be challenging. For example, facilities for management of clinical disorders in remote areas or evacuation to a lower altitude may be limited.
 CONSIDERATIONS OF EXPOSURE AND TIMECOURSE
CONSIDERATIONS OF EXPOSURE AND TIMECOURSE
Individual responses to hypobaric hypoxia may be conceptualized along a time continuum of interrelated phases: acute (immediate to 3–5 days in which acute illnesses present), subacute (over weeks leading toward acclimatization), chronic (years), and lifelong residence (Fig. 92-1). The reduction in environmental oxygen as a result of altitude exposure lowers the oxygen available for gas exchange in the lungs (PAO2), arterial oxygen (PaO2), and cellular oxidative phosphorylation for adenosine triphosphate (ATP) production.

Figure 92-1 Individuals respond to altitude with time-dependent acclimatization and illness expression, and populations living at altitude exhibit adaptations that differ according to population ancestry.
Oxygen levels in the atmosphere were extremely low; rose about 2.5 million years ago in the Great Oxidation event and then rose sharply.8 Over the past 600 million years, oxygen levels have varied from as low as ~12% to as high as ~30%.8,10 All multicellular animals11 have genetically encoded oxygen homeostasis pathways. We have a common ancestry and therefore share many elements of the response to hypoxia. People moved to live at high altitudes in the past 100,000 years and evolutionary forces have resulted in different strategies of adaptation in different populations.12
 RESPONSES TO HYPOBARIC HYPOXIA
RESPONSES TO HYPOBARIC HYPOXIA
Important adaptations to hypobaric hypoxia are considered below.
Ventilatory Adaptations
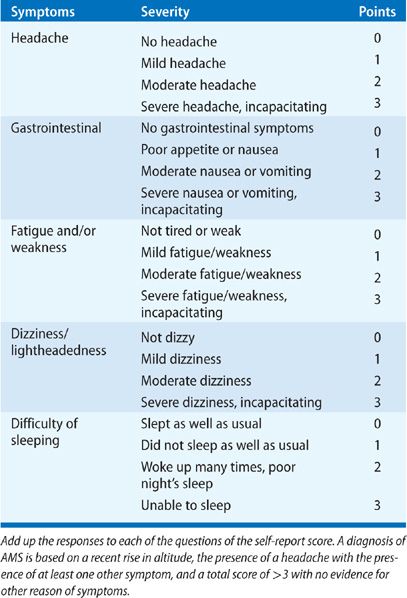
An increase in ventilation is an immediate vital, mammalian response. It results in a decrease in alveolar CO2 in order to increase PAO2, thus mitigating decreases in vascular oxygen delivery at altitude compared to sea level (Fig. 92-2).
Figure 92-2 Oxygen tension is lower along the axis of oxygen delivery at 4540 m or ~15,000 ft (red line) as compared with sea level (blue line). (Adapted from Hurtado A. Animals in high altitudes: resident man. In: Dill DB, ed. Handbook of Physiology. Section 4: adaptation to the Environment. Washington, D.C.: American Physiological Society;1964:843–859.)
The sensor initiating the acute response is the carotid body at the bifurcation of the internal and external carotid arteries. Ablation of the carotid body attenuates the acute response to altitude exposure.13
Blood flowing through the carotid body comes from the lungs via the carotid artery, where the oxygen pressure is typically 80 to 100 mm Hg. Carotid body afferent activity rises significantly as arterial oxygen pressure falls below 60 mm Hg.13 However, oxygen pressure in the mitochondria is much lower, as oxygen has to diffuse across vessels and interstitial spaces and is extracted by cells for a number of metabolic processes. In the microvessels of the carotid body, the average oxygen pressure is about 50 mm Hg with minimal values likely near 20 mm Hg. It is reasonable to expect an average mitochondrial oxygen pressure of about 40 mm Hg and a minimum of about 15 mm Hg. If this seems low, in the liver and spleen average tissue values are measured at less than 12 to 15 mm Hg.14 Thus, tissue in the carotid body may be exposed to oxygen pressure significantly less than typical levels of arterial oxygen pressure ~80 mm Hg.
Oxygen level is major driver of carotid body response although carotid body afferent activity can also be stimulated, at least transiently, by changes in carbon dioxide or hydrogen ion concentrations. Respiration is stimulated by decreasing the [ATP]/[ADP][Pi] ratio and, as a result, the rate of respiration does not decrease until oxygen deprivation is severe enough to cripple metabolism.14 Decreased availability of oxygen first affects the ability of mitochondria to maintain the [ATP]/[ADP][Pi] ratio, a measure of energy available when ATP is hydrolyzed to adenosine diphosphate (ADP) and inorganic phosphate (Pi). Mitochondrial ATP synthesis is tightly coupled to oxygen consumption; that is, mitochondria respire only fast enough to replenish ATP as it is used. However, other responses to lowered oxygen levels are engaged depending on the duration and severity of the oxygen deprivation. Cellular oxygen–sensor responses activate short- and long-term energy savings and cellular protection mechanisms.15
In the first few minutes of exposure, as PAO2 decreases to below ~60 mm Hg, ventilation increases, resulting in a decrease in PACO2.13,16 In the carotid body, low O2 and high CO2 interact synergistically to depolarize glomus cells and produce an action potential; the ventilatory effects of concurrent hypoxia and hypercapnia are greater than the sum of the two stimuli applied separately. As the PAO2 level declines, the relationship between the sensory afferent nerve activity and PACO2 becomes increasingly steeper, leading to higher ventilatory drive. The physiologic response to acute exposure is an abrupt increase in ventilation (Fig. 92-3), which continues to increase over days. After a few days adaptations take place in the brainstem controller through circuit adaptations so that ventilation is increased with reductions in PACO2 at all levels of PAO2, and the afferent responses from the carotid body also reflect this change in responsiveness to chemoreception even when there is a near restoration of pH. Even though this organ’s response is pathognomonic of the acclimatization process in response to altitude-associated chronic hypoxia, there is bidirectional, efferent as well as afferent, communication so that attributing the entire physiologic response to the carotid body is problematic.
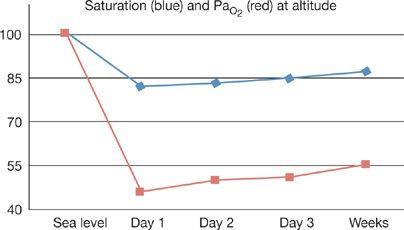
Figure 92-3 The figure illustrates the problem with hypoxemia encountered with acute altitude exposure. Values of arterial oxygen saturation (blue, % O2 saturation) and arterial oxygen tension (PaO2) before, during the first few days at altitude, and after weeks of residence.
Hypoxic chemotransduction in the carotid body requires release of excitatory transmitters that activate afferent sensory neurons.17 Cellular events including bioenergetic, biosynthetic, and conformational cause the chemotransduction.18 Mechanisms implicated include oxygen-sensitive ion channels, reactive oxygen species and the “redox” state of the cell, and direct effect of oxygen to alter the conformation of a protein, such as NADPH oxidase 4, an enzyme encoded by the NOX4 gene. This phagocyte-type oxidase has been postulated as an oxygen sensor of sorts because its activation is linearly and steeply dependent on oxygen availability. A cascade of immediate intracellular effects will initiate changes in afferent information from the carotid body to the medulla that will lead to the increased tidal volume and respiratory frequency.19
A physical remodeling of ventilatory control begins soon after exposure through changes in gene expression. In blood vessels this includes growth factors such as vascular endothelial growth factor (VEGF). The main pathway for such hypoxic responses is mediated by the hypoxia-inducible transcription factor 1 alpha (HIF-1α).15 HIF-1α is the oxygen-dependent subunit of the HIF-1 heterodimer, considered the master regulator of oxygen homeostasis that initiates transcription of over 100 hypoxia-inducible genes, that have hypoxia response elements. Under normoxia, HIF-1α is formed continuously and degraded in the cytoplasm. HIF-1α is hydroxylated by the enzyme prolyl hydroxylases prior to its degradation. The prolyl hydroxylases have a relatively high Km for oxygen; hydroxylation leads to degradation under physiologic conditions. Decreased oxygen pressure results in significant accumulation of HIF-1α in less than 2 minutes; HIF-1α is rapidly degraded upon reoxygenation.
Central controllers can be affected directly and indirectly by oxygen availability, but over time neurotransmitter expression correlates with changes in gain and changes in gene expression with some additive epigenetic changes.20 Within days of exposure, there is a progressive increase in sensitivity but a downregulation of the magnitude of the ventilatory response by the brainstem networks that integrate afferent information.21 At 4300 m, hyperventilation increases over the first few days and then over 1 to 2 weeks despite metabolic compensation to the respiratory alkalosis.22 The acclimatized person will continue to hyperventilate when returned to normoxia, a phenomenon not observed during the very first hour of hypoxic exposure. pH compensation to hypocapnia occurs in 2 to 4 days and a mild alkalinity persists over the following weeks. The ventilatory response to chronic hypoxia consists of both frequency and tidal volume changes. Acclimatized individuals will return to nearly normal control arterial acid–base status within 24 to 48 hours of normoxia, following acclimatization to 4300 m.
Thus, the transition points from acute to subacute to full acclimatization in the ventilatory domain blend from one to another.23 Acclimatization results from cellular and system responses that improve oxygen delivery. Ventilatory responses are complemented by systemic ones, including erythrocytosis and new blood vessel formation that further assist in increasing oxygen delivery to peripheral tissues. For instance, HIF-1α affects a variety of “hypoxic response elements” with downstream effects on many hypoxic response proteins including erythropoietin.24 Some actions of erythropoietin result in increases in hemoglobin levels, and thus will affect oxygen carrying capacity in the blood and an improvement in oxygen delivery, lasting weeks to months. People with a mutation limiting HIF-1 degradation have high baseline ventilation and exaggerated ventilatory response to hypoxia, as well as chronic polycythemia under nomroxia.25
 SLEEP AND PERIODIC BREATHING
SLEEP AND PERIODIC BREATHING
Healthy people at altitude may develop a pattern of regularly recurring periods of hyperpnea and apnea. This phenomenon results from a dynamic interaction among elements in two separate feedback loops, one through peripheral and the other through central chemoreceptor reflexes, with contributions by cardiac output and brain blood flow.26 Periodic breathing occurs during wakefulness but is especially evident in sleep, and apneas and hypopneas of the nonobstructive type are reported as occurring almost equally in non-REM and REM sleep in health subjects.27 Recurrent central apneas will occur during sleep at modest altitude (>2500 m), and is a manifestation of increased alveolar ventilation (hyperpnea) from hypoxia producing hypocapnia competing with a normal elevation in the apneic (carbon dioxide) threshold with sleep.28 The apneas or hypopneas produce a fall in arterial oxygen and a rise in carbon dioxide and lead to arousals from sleep during the hyperpneic phase.29 The alternating hyperpnea and apnea produce an oscillatory pattern of oxygen saturation, with the nadir of oxygen saturation playing some role in susceptibility to HAPE and HACE.30
At altitudes above 3000 m, the quality of both REM and non-REM sleep is impaired by arousals often associated with periodic breathing;31 at extreme altitude (above 5500 m) length of and number of apneas decrease due to a high frequency of breathing. The reason for this phenomenon could represent a greater relative influence of central oxygen–chemosensitive sites over afferent information on peripheral hypoxia and hypocapnia.32,33 With acclimatization, the sleep pattern tends to become more normal; however, periodic breathing during non-REM sleep may persist over a week or more.30
 THE PULMONARY CIRCULATION
THE PULMONARY CIRCULATION
Moderate-to-severe pulmonary hypertension (both systolic and diastolic) is a common finding in all the altitude-related diseases.4 Although pulmonary blood vessels are extensively supplied with sympathetic (vasoconstrictor) and parasympathetic (vasodilator) fibers, regulation of vasomotor tone is largely due to the local effects of oxygen and carbon dioxide. At altitude the entire lung is hypoxic and CO2 levels are low as a consequence of hyperventilation; as a result, altitude is a model of whole lung hypoxic, hypocapnic pulmonary vascular vasoconstrictive responses.4 Acute vasoconstriction over a few hours is a physiologic response and can be reversed as soon as the hypoxia is lifted. With days to weeks of hypoxic exposure there occurs a structural remodeling of pulmonary arterioles and an increase in resting pulmonary arterial pressure and pulmonary vascular resistance; hence pulmonary artery pressure does not decline immediately with restoration of normoxia. With return to sea level after weeks to months, right ventricular hypertrophy and pulmonary vascular pressures will return to normal over several weeks.4
Andean highlanders have high pulmonary artery pressures and do not restore pulmonary artery pressure or right ventricular hypertrophy to normal levels after returning to sea level or normal oxygen tensions. In contrast, Tibetan highlanders have minimal elevation of pulmonary artery pressure and do not exhibit the vesicular changes of the Andean highlanders.34
The pulmonary circulation is a high-flow, low-pressure system.35 Based upon a balance of hydrostatic and oncotic pressure gradients across pulmonary capillary microvessels, and by Na+ pumps, the pulmonary interstitium and alveolar spaces are maintained in a “dry” state. At high altitude, while respiratory mechanics do not change appreciably, changes in hemodynamics due to vasoconstriction and high shear stress are dramatic and in certain individuals lead to acute presentations of pulmonary edema.4
 FLUID HOMEOSTASIS
FLUID HOMEOSTASIS
Dermal edema is seen in faces of many, but not all mountaineers at high altitude36 and in the retinal vessels of almost all individuals.37 Lung edema, cerebral edema, and peripheral edema are the hallmarks of disease. Extravasation may also result from loss of tight channels due to oxidative stress and free radicals or a direct effect of hypoxia on ion channels.38 Local dysregulation of the renin–angiotensin–aldosterone system is proposed to play a role in this generalized response.39,40
 ERYTHROPOIESIS AND HEMOGLOBIN AFFINITY
ERYTHROPOIESIS AND HEMOGLOBIN AFFINITY
An increase in the number of red blood cells occurs with acute exposure owing to an increase in EPO synthesis in response to HIF-1 and HIF-2.41,42 This acclimatization response takes a week or so and is reversible upon return to low altitudes. With chronic hypoxia, there are an increased number of erythrocytes and an increase in oxygen content among upward migrants such as Colorado residents and among Andean highlanders. Tibetan highlanders have minimal or no erythrocytosis owing to the high frequencies of variants in EPAS1 (HIF-2α) associating with lower hemoglobin concentration.43
Several studies have considered the potential for a change in oxygen affinity. The increase in ventilation results in decreases in alveolar and arterial PCO2 and arterial [H+]; concomitantly, serum levels of 2,3-diphosphoglycerate (2,3-DPG) are increased. While the reductions in PaCO2 and [H+] increase hemoglobin affinity for O2, increases in 2,3-DPG diminish the affinity; loading and unloading of O2 from hemoglobin depends upon the balance of these factors.44 Humans, native to the Andes or Himalayan mountains, also can increase O(2) affinity, not because of a fundamental difference in hemoglobin structure or function, but because of extreme hyperventilation and alkalosis.45
COMMON CLINICAL DISORDERS OF HIGH ALTITUDE
Altitude-related clinical disorders are well recognized. They may be observed occasionally at altitudes as low as 8000 ft (2500 m), however their frequency increases with increasing altitude. A recent clinical practice review is available from the Medical Commission of International Mountaineering and Climbing Federation (UIAA).46 Information about high-altitude travel for those with pulmonary disease is also available.47
 HIGH-ALTITUDE HEADACHE
HIGH-ALTITUDE HEADACHE
High-altitude headache (HAH) is very common, and is exacerbated by insufficient hydration in the setting of increased water loss with hyperventilation, overexertion, and insufficient energy intake, particularly in those who have experienced HAH on a previous visit to altitude. Vasodilation from hypocapnia may also contribute, but there is little evidence for brain edema in those without neurologic symptoms.48,49
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


