Figure 8.1
Suggested risk stratification for life threatening cardiac events in non- genotyped patients with the long QT syndrome. ACA aborted cardiac arrest, KM Kaplan Meier, SCD sudden cardiac death. The figure is taken with permission from reference2
Risk stratification among genotyped LQTS patients can be based on genotype-specific factors found to affect the phenotypic expression in patients with LQTS; those risk factors include age, gender, the post partum time period, menopause, prior syncope, mutation location, type of mutation (missense/non-missense), the biophysical function of the mutation and response to betablockers [13, 14]. Figures 8.2 and 8.3 show suggested risk stratification schemes for patients with LQT1 and LQT2, respectively.
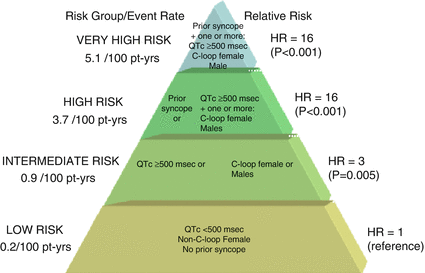
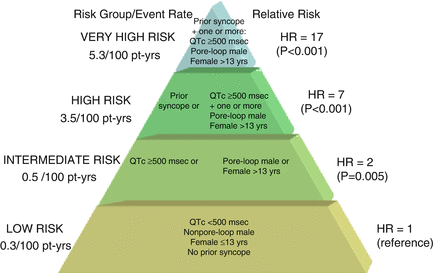
Figure 8.2
Proposed risk stratification for aborted cardiac arrest or sudden cardiac death in LQT1. C-loop mutations in cytoplasmic loops of the KCNQ1 channel, Pt-yrs patient years, HR hazard ratio. The figure is taken with permission from reference47
Figure 8.3
Proposed risk stratification scheme for aborted cardiac arrest or sudden cardiac death in LQT2 patients. Pore-loop muations in the pore-loop segments of the KCNH2 (hERG) channel, Pt-yrs patient years, HR hazard ratio. The figure is taken with permission from reference14
The rare forms of LQTS Jervell-Lange-Nielsen syndrome (autosomal recessive inheritance form of LQTS) and Andersen-Tawil syndrome (LQTS type 7) are both associated with very poor prognoses (unless ICD is implanted); Patients with these syndromes experience life threatening arrhythmic events at an early age. Similarly, patients with multiple LQTS-associated mutations, particularly double mutations affecting the same gene, have been associated with a greater risk for life threatening arrhythmic events than patients who harbor a single mutation [15].
Management
In general, the treatment of LQTS consists of lifestyle modifications, medical therapy with beta-blockers, ICD and/or surgical therapy. The ACC/AHA/ESC guidelines [16] and a recently published expert consensus statement [3] recommend lifestyle modifications for all patients with a diagnosis of LQTS. Beta-blockers are recommended as a Class I indication for all patients with a clinical diagnosis of LQTS and as a Class IIa indication for patients with a genetic diagnosis of LQTS who have a normal QTc duration. Although there are limited data on the most effective dosage of beta-blockers, full dosing adjusted for age and weight is recommended. Abrupt discontinuation of beta-blockers should be avoided as this may cause exacerbation [3]. Implantation of an ICD is recommended for LQTS patients who experience an aborted cardiac arrest (class I indication) or for patients who had syncope and/or VT despite beta-blockers therapy (class IIa indication). The recently published expert consensus statement [3] recommends performing left cardiac sympathetic denervation (LCSD) in high-risk patients with a diagnosis of LQTS in whom ICD therapy is contraindicated or refused and/or beta-blockers are either not effective in preventing syncope/arrhythmias, not tolerated, or contraindicated (class I indication). In addition, the consensus statement has added that sodium channel blockers can be useful, as add-on therapy, for LQT3 patients with a QTc >500 ms who shorten their QTc by >40 ms following an acute oral sodium channel blocker test (class IIa indication) [3].
Lifestyle Modifications
The fact that patients with certain genotypes are more likely to experience their events under well-defined circumstances may provide insights into preventive measures. Patients with LQT1 have most of their events during exercise. Therefore, they should avoid strenuous exercise activity (particularly swimming) without supervision, and those at intermediate or high risk should not engage into competitive sports [3, 16]. Patients with LQT2 should be advised to avoid unexpected auditory stimuli as their cardiac events are predominantly associated with sudden arousal [17, 18]. Removal of loud noise stimuli at home and work such as elimination of alarm clocks, door bells and telephone ringing is usually recommended. LQT3 patients mainly experience events during sleep and at rest, and should be considered for a special intercom system in the bedroom. All patients with LQTS should avoid drugs known to prolong QT interval, or affect potassium and magnesium level. It is important to identify and correct electrolyte abnormalities that may occur during diarrhea, vomiting, metabolic conditions, or imbalanced diets for weight loss [3].
Beta-Blockers
Beta-blocker therapy is the mainstay treatment of patients with LQTS. The efficacy of this therapy in LQTS has been demonstrated in multiple studies. Moss et al. [19] have reported the efficacy of beta-blockers in 869 LQTS patients. Beta-blocker therapy was associated with a significant reduction in the rate of cardiac events in probands (0.97 ± 1.42 to 0.31 ± 0.86 events per year, p < 0.001) and in affected family members (0.26 ± 0.84 to 0.15 ± 0.69 events per year, p < 0.001). In another study among 549 LQT1 and 422 LQT2 patients from the International LQTS Registry, we have found that Beta-blocker therapy was associated with a prominent risk-reduction in high-risk patients, including a 67 % reduction (P = 0.02) in LQT1 males and a 71 % reduction (P < 0.001) in LQT2 females [20].
The protective effects of beta-blockers among LQTS patients may also depend on mutation location. We have shown among 860 patients with genetically confirmed LQT1 that beta-blocker therapy was associated with a significant 88 % reduction in the risk of life-threatening cardiac events among LQT1 carriers of the cytoplasmic loops (C-loop) missense mutations (p = 0.02), whereas among LQT1 carriers of non-C-loop missense mutations there was no significant reduction in the risk for life threatening cardiac events (HR 0.82, p = 0.68) [21]. It is known that the C-loops play an important role in the sympathetic regulation of the KCNQ1 channel [22]. Cellular expression studies have suggested that there is a combination of decrease in basal function and altered adrenergic regulation of the IKs channel in patients with C-loops missense mutations that may provide a potential explanation why beta-blockers are particularly effective in patients with this type of mutation [21].
Potassium Supplementation
Potassium supplementation and spironolactone were proposed for patients with LQT2 who exhibit mutation of the KCNH2 gene. KCNH2 function is highly dependent on the extracellular potassium. It has been suggested that potassium administration will increase serum potassium level and improve repolarization abnormalities. Two small studies have shown that potassium supplements and spironolactone are associated with a significant shortening of the QTc [24, 25]. Unfortunately, there are no data that potassium supplements or spironolactone can decrease the risk of cardiac events.
Sodium Channel Blockers
Over the last decade, sodium channel blockers such as mexilitine and flecainide have been investigated as a potential treatment option for patients with LQT3. Both Mexilitine [26] and Flecainide [27–31] administration are associated with significant shortening of the QT interval among LQT3 patients.
Technical Aspects
QTc values
The normal and prolonged QTc values depend on age and gender. Suggested QTc values for diagnosing QTc prolongation are: QTc >460 ms during childhood (ages 1–15 years), QTc >450 ms for adult males, and QTc >470 ms for adult females [32].
QT and QTc measurement
The QT interval should be determined as a mean value derived from at least 3–5 cardiac cycles, and is measured from the beginning of the QRS complex to the end of the T wave.
The QT measurement should be made in leads II and V5 or V6, with the longest value being used. The main difficulty lies when there are T and U waves that are close together. When T-wave deflections of a near-equal amplitude result in a biphasic T wave, the QT interval is measured to the time of final return to baseline. If a second low amplitude repolarization wave interrupts the terminal portion of the T wave, it is difficult to determine whether the second deflection is a biphasic T wave or an early-occurring U wave. In such cases, it is best to record both the QT (measured at the end of the first deflection) and the QTU (measured at the end of the second deflection) intervals [32].
The Bazett formula is widely used to correct the QT interval for heart rate (QTc); QTc equals QT divided by the square root of the R-R interval (all intervals should be measured in seconds).
Epinephrine QT stress test
This provocative test may aid in unmasking individuals with concealed LQT1 [8]. There are two available protocols: by bolus infusion (Shimizu protocol) or an incremental, escalating infusion (Mayo protocol). According to the Mayo protocol [8], after 10 min of rest, 12 lead ECG recording speed was set at 50 mm/s, baseline parameters were obtained (including QT and QTc), and then an infusion of epinephrine was initiated at 0.025 μg/kg/min. After 10 min of the infusion, the measurements were repeated. The epinephrine infusion was then increased sequentially to 0.05, and 0.1 μg/kg/min, and the measurements were repeated 5 min after each dose increase. The epinephrine infusion was then discontinued, and measurements were obtained 5 and 10 min afterwards. A paradoxical response characterized by uncorrected QT lengthening (ΔQT ≥30 ms) rather than expected shortening appears diagnostic for LQT1 (with a sensitivity and specificity of 92 and 86 %, respectively).
Brugada Syndrome
Incidence and Etiology
Brugada syndrome is another familial disorder with structurally normal heart that involves mutations in genes encoding myocyte ion channels. Brugada syndrome is characterized by a specific ECG pattern of coved-type ST-segment elevation in the right precordial leads (V1 through V3) accompanied by a susceptibility to polymorphic VT and SCD [33]. Brugada syndrome prevalence has been estimated at 1 per 2,000 people worldwide. The prevalence is higher in Southeast Asian countries, especially Thailand, Philippines and Japan [34, 35].
Brugada syndrome is typically inherited through an autosomal dominant mode of transmission. To date, 12 Brugada syndrome-associated genes have been reported [35], with all mutations leading to either a decrease in the inward sodium or calcium current or an increase in outward potassium current.
Approximately 25 % of cases of Brugada syndrome result from mutations in the SCN5A gene that encodes for the α subunit of the cardiac sodium channel. Overall, the genetic cause has been identified for only 30 % of clinically diagnosed Brugada syndrome patients.
Diagnosis
Three ECG patterns associated with Brugada syndrome were described.
Type 1 is characterized by a J point elevation ≥2 mm (0.2 mV), a coved ST-segment elevation followed by a negativeT wave. This ECG pattern is diagnostic of Brugada syndrome. Type 2 has a J point elevation ≥2 mm, ST-segment elevation has a saddleback appearance, and then either a positive or biphasic T wave. Type 3 has either a saddleback or coved appearance with a J point elevation <2 mm, and an ST-segment elevation of <1 mm.
Type 2 and type 3 ECG are not diagnostic of the Brugada syndrome.
According to the recently published expert consensus statement [3] Brugada syndrome is definitively diagnosed when a type 1 ST-segment elevation is observed either spontaneously or after intravenous administration of a sodium channel blocking agent in at least one of the precordial leads V1 and V2, which are placed in the 2nd, 3rd or 4th intercostal space.
Brugada syndrome is diagnosed in patients with type 2 or type 3 ST-segment elevation in ≥1 lead among the precordial leads V1, V2 positioned in the 2nd, 3rd or 4th intercostal space when intravenous administration of a sodium channel blocking agent induces a type I ECG pattern.
Due to the low diagnostic yield of genetic testing among clinically diagnosed Brugada syndrome patients (genetic abnormalities are found in about 30 %), genetic testing is not recommended in the absence of a diagnostic ECG. Genetic testing may be useful for family members of a successfully genotyped proband.
Prognosis and Risk Stratification
Brugada syndrome typically manifests during adulthood, with ventricular tachyarrhythmic events occurring at an average age of 40 years and sudden death typically occurring during rest or at sleep [34]. Brugada syndrome is 8–10 times more prevalent in males than in females. At the time of diagnosis, males are more likely than females to present with previous symptoms, a spontaneous type I ECG pattern, and VF during an electrophysiology study (EPS) [36]. Although the basis for this sex-related distinction is unknown, it has been suggested that there is some sexual differences in gene expression or function and that the more prominent potassium transient outward current (Ito) in males may contribute to the male predominance of the syndrome [37].
Currently, risk stratification in Brugada syndrome relies mainly on clinical factors. The most important clinical risk factor associated with high risk of life threatening arrhythmias is the presence of syncopal episodes in patients with a spontaneous type I ECG. Other risk factors include the presence of fragmented QRS, spontaneous atrial fibrillation, and ventricular effective refractory period <200 ms during an EPS [38].
There is controversy regarding the value of programmed electrical stimulation during an EPS in predicting the risk for cardiac events. Brugada et al. [39] have found that inducibility of ventricular arrhythmias is a marker of a poor prognosis by multivariate analysis, but in both the PRELUDE (Programmed electrical stimulation predictive value) [38] and the FINGER (France, Italy, Netherlands, GERmany) [40] registries, inducibility of sustained ventricular arrhythmias did not predict ventricular arrhythmic events by multivariate analysis. Family history of SCD was not found to be an independent risk factor for cardiac events.
There are limited data on how knowledge of the Brugada syndrome specific mutation may assist in risk stratification. The presence of an SCN5A mutation has not been proven to be a risk marker in Brugada syndrome, but non-missense mutations that result in a truncated protein or missense mutations with greater than 90 % INa reduction have been found to predict syncopal episodes [41].
Management
The treatment of Brugada syndrome consists of lifestyle changes, ICD implantation and possibly medical therapy with Quinidine.
The expert consensus statement [3] recommends three lifestyle modifications for all patients with a diagnosis of Brugada syndrome: (1) avoidance of drugs that may induce or aggravate ST-segment elevation in the right precordial leads (avoiding some Ia and Ic antiarrhythmic drugs, several psychotropic drugs, and several anesthetics/ analgesics including Propofol); (2) avoidance of excessive alcohol intake; and (3) immediate treatment of fever with antipyretic drugs.
ICD implantation is recommended for patients with a history of aborted cardiac arrest or spontaneous sustained VT for secondary prevention of SCD (class I indication). ICD can be useful for patients with a spontaneous type I ECG who have a history of syncope believed to result from VT. (Class IIa indication)
ICD implantation may be considered (Class IIb indication) in patients with Brugada syndrome and inducible VF during EPS.
Quinidine can be useful in patients who qualify for an ICD but have a contraindication for ICD implantation or refuse implantation. (Class IIa)
Quinidine may be considered (Class IIb) in asymptomatic patients with Brugada syndrome who have spontaneous type I ECG.
Patients with Brugada syndrome who experience arrhythmic storms (2 or more VT/VF episodes in 24 h) may be treated by quinidine, Isopreterenol (class IIa) or by catheter ablation targeting fractionated right ventricular electrograms (class IIb).
Technical Aspects
Procaineamide provocative testing
This provocative test may aid in unmasking individuals with Brugada syndrome. Baseline ECG (including high precordial leads) and vital signs should be taken at baseline, than start IV Procainamide 1 g over 30 min, measure vital signs and perform ECG every 10 min, stop infusion at 30 min and repeat ECG and vital signs at 40, 60, and 90 min. When performing provocative testing with a sodium channel blocker, the infusion should be terminated when a type-1 ECG develops, premature ventricular beats or other arrhythmias develop, or the QRS widens to ≥130 % of baseline [42].
Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia (ARVC/D)
Incidence and Etiology
ARVC/D is a genetic heart muscle disease; its true prevalence is unknown, with estimates between 1 in 2,000 and 1 in 5,000. It is characterized pathologically by fibrofatty replacement of the right ventricular (RV) myocardium. The resulting disruption of the RV myocardial architecture in ARVC/D can lead to RV dysfunction, ventricular tachyarrhythmias, syncope, or SCD [34].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


